End-to-End Manufacturing of Biosimilars – From Cell Line to Commercial Product
- Biosimilars are complex biological medicines that require a sophisticated development and manufacturing process.
- An end-to-end approach covers every step from the initial gene sequence to the final vialed product.
- This integrated pathway ensures that the biosimilar is developed efficiently while meeting strict quality and regulatory standards.
What Does End-to-End Manufacturing Mean in Biosimilars?
End-to-end manufacturing in the context of biosimilars refers to a fully integrated development and production cycle that spans from the genetic blueprint of the molecule all the way to the filled and finished drug product. In practical terms, it means coordinating every phase as one continuous workflow. By covering biosimilars process development holistically, end-to-end manufacturing ensures that each stage is aligned with the next, reducing gaps or inefficiencies.
This approach is often provided by specialized CDMO companies that offer “gene-to-vial” services. Such providers handle everything under one roof, which can shorten timelines and simplify management. For example, an integrated service can take a biosimilar from cell line development to GMP manufacturing in a seamless manner, ready for first-in-human studies. In contrast, a non-integrated approach might involve multiple vendors or hand-offs between different teams, which can introduce delays or inconsistencies.
A key feature of end-to-end biosimilar manufacturing is rigorous alignment with quality and regulatory requirements at every step. Because biologics are sensitive and highly regulated, having one integrated pipeline helps maintain control. Each phase is executed with the final goal in mind: producing a biosimilar that is highly similar to the reference biologic in terms of safety, purity, and potency. An end-to-end strategy facilitates this by ensuring all development decisions (from cell engineering to purification methods) contribute to that ultimate comparability goal.
Crucially, an end-to-end model also emphasizes cross-functional collaboration. Process scientists, cell line engineers, and quality experts work in concert rather than in isolation. This means feedback from later stages (for example, analytical characterization) can loop back to earlier stages (like cell line or process optimization) in real time. The result is a more efficient biosimilars manufacturing cycle.1
Indeed, companies adopting fully integrated workflows often report faster development times and fewer costly surprises. They can accelerate innovative therapies and reduce time to market while upholding the highest quality standards.
How Is a Biosimilar Cell Line Developed and Optimized?
Developing an optimal biosimilar cell line is the foundation of success, as the cell line that produces the therapeutic protein. The process starts with the gene that encodes the target biologic. Scientists insert this gene into a suitable host cell (commonly CHO cells for monoclonal antibodies development, due to their robustness and human-like protein processing). These cells are then cultured and screened to identify clones that produce the protein in high quantity and with the correct quality attributes.
Cell line development for biosimilars must achieve more than just high productivity. It must also ensure that the protein produced is highly similar to the originator’s product. This means the cell’s protein folding, modification, and glycosylation patterns should match the reference product as closely as possible. Achieving this often involves evaluating many clones. Each clone might produce slightly different variants of the protein, so developers use extensive analytical characterization to select a clone that yields a product with the desired similarity in critical quality attributes (CQAs) such as glycan profiles, charge variants, and bioactivity.2
Once promising high-producing clones are identified, further optimization is undertaken. This includes refining the culture conditions and genetic stability of the cell line. For instance, scientists optimize the growth media composition, feeding strategy, and other culture parameters to maximize expression levels without compromising product quality. The goal is a robust and high-performing producer cell line that consistently expresses the target protein with the right attributes. Over time, the selected cell line undergoes stability studies to ensure it maintains performance over many generations. It is a critical aspect since manufacturing of biosimilars may continue for years. The developed cell line is then banked to secure its use for the entire product lifecycle. Master Cell Banks (MCBs) and Working Cell Banks (WCBs) are created under stringent conditions.
Developing and optimizing a biosimilar cell line is an exercise in precision engineering of living cells. When done successfully, the result is a stable cell line capable of consistently manufacturing the biosimilar at scale, with each cell acting as a tiny factory churning out molecules nearly indistinguishable from those of the original biologic drug. This cell line then becomes the basis for all subsequent manufacturing steps.
Key Steps in Upstream and Downstream Process Development
During upstream development, scientists design the bioreactor conditions and media to optimize cell growth and productivity. The cells might be grown in large stainless-steel tanks or single-use bioreactors under carefully controlled conditions (temperature, pH, oxygen, nutrient feed rates). The goal is to achieve high cell densities and high protein titers while maintaining product quality. Variables such as the composition of the growth medium, feeding schedules, and cultivation time are systematically optimized.3 For manufacturing of biosimilars, upstream process development is particularly attentive to consistency. A well-optimized upstream process will yield a high volume of the target protein while controlling variants and ensuring the cells remain healthy throughout the run.
Downstream process development is where the raw harvest is transformed into a purified drug substance. This phase is multi-step and highly engineered. It typically begins with a capture step, often using affinity chromatography. For example, monoclonal antibody biosimilars commonly use Protein A affinity chromatography as the first purification step, which specifically binds antibodies and separates them from other components. This capture step drastically reduces impurities such as host cell proteins and DNA, concentrating the product and providing a cleaner starting point for further purification.4 Following the initial capture, additional polishing steps are employed, such as ion-exchange chromatography, which remove remaining trace impurities by separating molecules based on charge, and perhaps hydrophobic interaction or mixed-mode chromatography targeting other contaminants.
Another critical element of downstream processing for biologics is viral clearance. Because biologics are produced in living cells, regulators require that manufacturers demonstrate the process can remove or inactivate potential viral contaminants. Techniques such as low-pH viral inactivation (exposing the product to acidic conditions to inactivate viruses) and nanofiltration (filtering out virus-size particles) are integrated into the downstream workflow. These steps ensure the biosimilar is safe from adventitious viruses.
Ultrafiltration/diafiltration steps then concentrate the protein and exchange it into the desired formulation buffer. Each downstream unit operation – be it a chromatography column, a filter, or a centrifuge – is optimized to maximize yield and purity while maintaining the protein’s integrity. The combined effect is a purification train that yields a drug substance of high purity, often over 99%, with impurity levels (like host cell protein or DNA content) well below regulatory limits. It is during downstream development that the biosimilars purification process truly defines the final product’s quality. As one industry resource emphasizes, without effective downstream purification, modern biologics manufacturing cannot meet the required standards for safety and efficacy.
From Lab to Plant – Why Tech Transfer and Scale-Up Are Critical?
Developing a successful process in the laboratory or pilot scale is only half the battle; the next major challenge is scaling it up to commercial production. Tech transfer refers to the systematic process of transferring a developed process (and all its associated knowledge) from the development environment (lab or pilot plant) to the production environment (manufacturing plant). Scale-up is the act of increasing the batch size – for example, moving from a 5-liter lab bioreactor to a 2,000-liter production bioreactor – while aiming to achieve the same product quality and yield. In biosimilar manufacturing, effective tech transfer and scale-up are absolutely critical, because any inconsistencies introduced at this stage can impact the biosimilar’s similarity to the reference product and its overall viability.
One key reason tech transfer is so important is the inherent sensitivity of biological processes. A bioprocess that works in a small controlled setting can behave differently at manufacturing scale. Physical differences, such as mixing efficiency, oxygen transfer, and temperature uniformity, emerge in larger bioreactors. These changes can affect cell growth or protein quality if not properly understood and managed.
Ensuring biosimilarity is a multifaceted challenge because biological systems are inherently variable, and thus achieving comparability with the reference product requires addressing several important aspects throughout the development and approval process.
In practice, this means scaling parameters like stirring speed, aeration rate, and feeding profiles must be carefully adjusted so that the cells feel as similar as possible between the lab and the plant. Engineering runs and scale-down models are often used to predict and troubleshoot scale-related issues before full production.5
Tech transfer is a highly disciplined project. It involves detailed documentation of the process and often a collaboration between the development scientists and the manufacturing engineers. The receiving production team needs to understand the rationale behind each process parameter and critical control point. Scale-up amplifies any small issues. If a slight fluctuation in pH was observed in a 5 L experiment, in a 5,000 L tank that fluctuation could be larger if not controlled. Therefore, the process development must have built-in robustness. The concept of process robustness and consistency becomes very tangible in the manufacturing of biosimilars. During tech transfer, aspects like equipment differences are addressed: for instance, the lab might use one type of centrifuge or filter, whereas the plant uses a different model. The process is adjusted to fit the production equipment and facility while preserving the process intent and product quality. This phase may include demonstration or engineering batches at intermediate scales to ensure everything performs as expected. 6
Last but not least critical aspect of scale-up is ensuring supply chain and materials remain consistent. The cell culture media, resins, filters and other consumables used in the lab must have equivalents at scale that perform the same. A stable supply of these high-quality raw materials is necessary to avoid variability. Tech transfer teams work on qualifying vendors and materials for large-scale lots and often manufacture validation batches to show that the large-scale process produces product equivalent to the lab batches.
Benefits of an Integrated End-to-End Approach in Biosimilar Production
An end-to-end tech transfer approach offers seamless continuity from lab development to manufacturing, which better meets a biotech company’s needs. The same provider can maintain uniform process parameters, equipment platforms, and materials throughout development and scale-up, which improves reproducibility. By using a single integrated provider, all process knowledge and data are transferred internally without the loss of critical information that often occurs when handing off to a different organization. A unified team (as both the “sending” and “receiving” unit are within one company) ensures that development insights carry through to scale-up, avoiding miscommunications. There is no need to manage multiple vendors or duplicate tech transfer activities, which reduces delays and costs associated with coordinating two different providers. This continuity minimizes knowledge gaps and surprises during scale-up, making it easier to identify and address potential issues early — a critical success factor in biosimilars production.
Additionally, a single-provider strategy streamlines coordination and logistics for the biotech company. Instead of negotiating separate contracts and aligning different organizations for development vs. manufacturing, the company can rely on one partner, which means fewer supplier agreements and a simpler supply chain to manage. This reduction in complexity is especially valuable for resource-limited biotech firms, as it frees them from having to orchestrate communication between two independent service providers. An integrated CDMO takes on the burden of internal tech transfer between its own departments, sparing the sponsor from that task and ensuring know-how is efficiently shared in-house. In essence, the end-to-end approach provides a one-stop solution that is more efficient and aligned with the biotech company’s timeline and budget.
Download our free e-book, which transparently shows how modular CDMO partnerships accelerate biopharma progress.
Challenges and Risks in End-to-End Biosimilar Manufacturing
While an end-to-end approach offers many benefits, it does not eliminate the inherent challenges and risks in biosimilar manufacturing. In fact, the very nature of biologics and the complexity of mirroring an existing product mean that developers must navigate numerous obstacles.
It’s worth noting that biosimilar programs operate under significant economic pressures. Development costs are high due to the regulatory complexities, and delays or failures can be financially devastating. Because multiple companies may pursue biosimilars of the same blockbuster biologic, there is often a race to market; this puts pressure on timelines and can tempt shortcuts, which are risky. Choosing a CDMO with a proven track record increases the chances of bringing the project to market first. It’s a key advantage in biosimilar production.
Even with careful development, scaling and maintaining a biologics process is fraught with technical challenges. Certain biosimilars, especially more innovative ones like bispecific antibodies or fusion proteins, might demand additional process steps (for example, extra purification or refolding steps), adding complexity and points of potential failure. Ensuring each unit operation is validated and works in tandem with others is like maintaining a complex machine. Any part failing can halt the entire production.7
The regulatory landscape for biosimilars is stringent and evolving. Regulatory agencies examining a biosimilar application look for a coherent story of development, where each part of the CMC package supports the next. Guidelines continue to be refined as more biosimilars are approved, which can shift expectations mid-development. A unified end-to-end development generates a comprehensive and well-aligned documentation trail. All data – from initial clone selection studies to process validation and lot release – can be compiled in a consistent format, making it easier to prepare regulatory submissions. Such a situation occurred in 2025, when both the EMA and FDA decided that the approval process for biosimilar drugs could be shortened. The implementation of the Streamlined Biosimilar Drug Development approach and the waiver of Clinical Efficacy Studies for biosimilars significantly reduced the time to market and potentially lowered costs for manufacturers.8
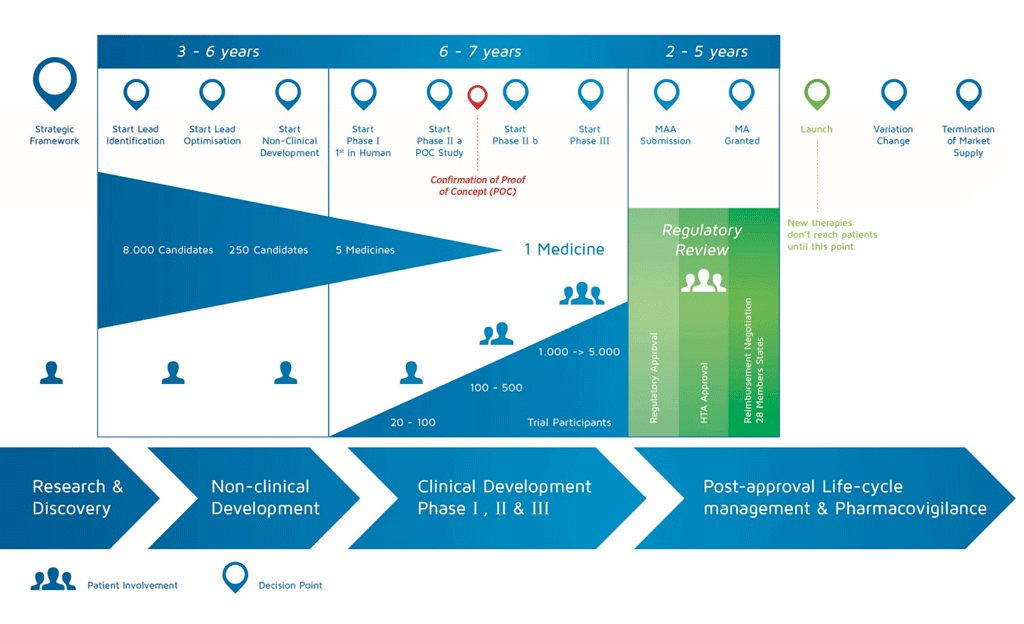
A unique challenge to biosimilars is the mandate to prove comparability not just once, but continuously. It’s not enough to match the reference product in initial characterization; the biosimilar must remain consistent in every batch produced. Manufacturers implement ongoing comparability protocols to make sure each production lot is within the tight specifications that were approved. Even after commercial launch, changes in the process until 2025 required comparability studies to ensure the biosimilar remains essentially the same. This need adds a layer of risk. Any drift in product attributes might require investigation and could potentially lead to product recalls or market withdrawal if not resolved. The necessity of rigorous quality control at all times cannot be overstated.
Conclusion
End-to-end development has emerged as a strategic model for bringing biosimilars from concept to commercial reality. By integrating all development stages this approach aims to ensure efficiency, quality, and compliance throughout the journey. It empowers companies to reduce time-to-market and better control their product’s destiny. However, success in biosimilars manufacturing still demands deep expertise, careful planning, and constant attention to detail. For decision-makers steering biosimilar programs, understanding the intricacies of this end-to-end process is crucial.
FAQ
Prepared by:

Marketing Specialist
References
- Partopour B, Pollard D. Advancing biopharmaceutical manufacturing: economic and sustainability assessment of end-to-end continuous production of monoclonal antibodies. Trends Biotechnol. 2025; 43(2): 462-475.
- Majumdar S, Desai R, Hans A, Dandekar P, Jain R. From Efficiency to Yield: Exploring Recent Advances in CHO Cell Line Development for Monoclonal Antibodies. Mol Biotechnol. 2025; 67(2): 369-392.
- Ranbhor R. Advancing Monoclonal Antibody Manufacturing: Process Optimization, Cost Reduction Strategies, and Emerging Technologies. Biologics. 2025; 19: 177-187.
- Mazzer AR, Perraud X, Halley J, O’Hara J, Bracewell DG. Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. J Chromatogr A. 2015; 1415: 83-90.
- Challener CA. Overcoming Biosimilar Scaling Challenges. Pharm. Technol. 2025; 49(1): 20-22.
- Judd N, Liu M, Nogal N. Considerations for a successful tech transfer of a biologics upstream process. Eur. Pharm. Rev. 2024; 5.
- Sinha S, Raphael R. Developing Biosimilars: Challenges and Opportunities. Pharmaceut Med. 2025; 39(5): 341-352.
- Tuszyner A. The New Era of Biosimilar Development: Seizing the Opportunity Under EMA’s Streamlined Guidelines. Mabion Science Hub. 2025


