Biologics Characterization for Ensuring Product Quality and Consistency
Analytics, Biologics
- Biologics characterization is the cornerstone of understanding molecular identity, structure, and function in complex biologic therapeutics. It ensures every product batch meets predefined quality, purity, and potency standards required by global health authorities.
- Mabion Biologics CDMO applies comprehensive analytical platforms covering all biologic modalities from gene to vial. This integrated approach guarantees biologics consistency, enabling safe and effective transition from research to clinical application.
- Regulatory agencies require detailed biologics characterization to confirm identity, purity, and potency. Mabion designs its analytical framework to exceed international standards like ICH Q6B and EMA guidelines. Early and thorough characterization supports efficient regulatory submissions and accelerated clinical readiness.
What Is Biologics Characterization and Why Is It Essential
Biologics characterization refers to the comprehensive analysis of biological drug products to determine their molecular and product attributes. Biologics are produced in living cells and are inherently more complex and variable than small molecule drugs. Thorough characterization is essential for understanding a biologics structure, composition, and functional properties, ensuring that each batch of product is of the intended quality. As a CDMO working with diverse biologic modalities like monoclonal antibodies, bispecifics, fusion proteins, antibody-drug conjugates, etc., Mabion Biologics CDMO emphasizes extensive characterization from gene to vial to establish a strong foundation for product development and safety.
Biologics exhibit inherent molecular heterogeneity and variability due to their production in living systems. Even minor changes in cell culture or process conditions can introduce subtle differences in a biologic’s glycosylation patterns, charge variants, or higher-order structure. Characterization is therefore critical to identify and control these variants before they impact clinical performance.1 By profiling attributes such as amino acid sequence, post-translational modifications, and impurity levels in great detail, developers gain deep product knowledge. This knowledge allows for setting appropriate specifications and process controls so that the final product consistently meets its identity, purity, potency, and safety requirements.
Rigorous biologics characterization is also mandated by regulatory authorities as part of biologics development. We integrate characterization early in development to guide process design and ensure each new therapeutic protein is well-understood prior to clinical trials. This proactive approach mitigates risks by revealing any critical quality attributes that could affect efficacy or immunogenicity.2
Key Analytical Methods Used in Biologics Characterization
Characterizing biologic products requires a broad arsenal of analytical methods, each probing different aspects of the molecule’s attributes.
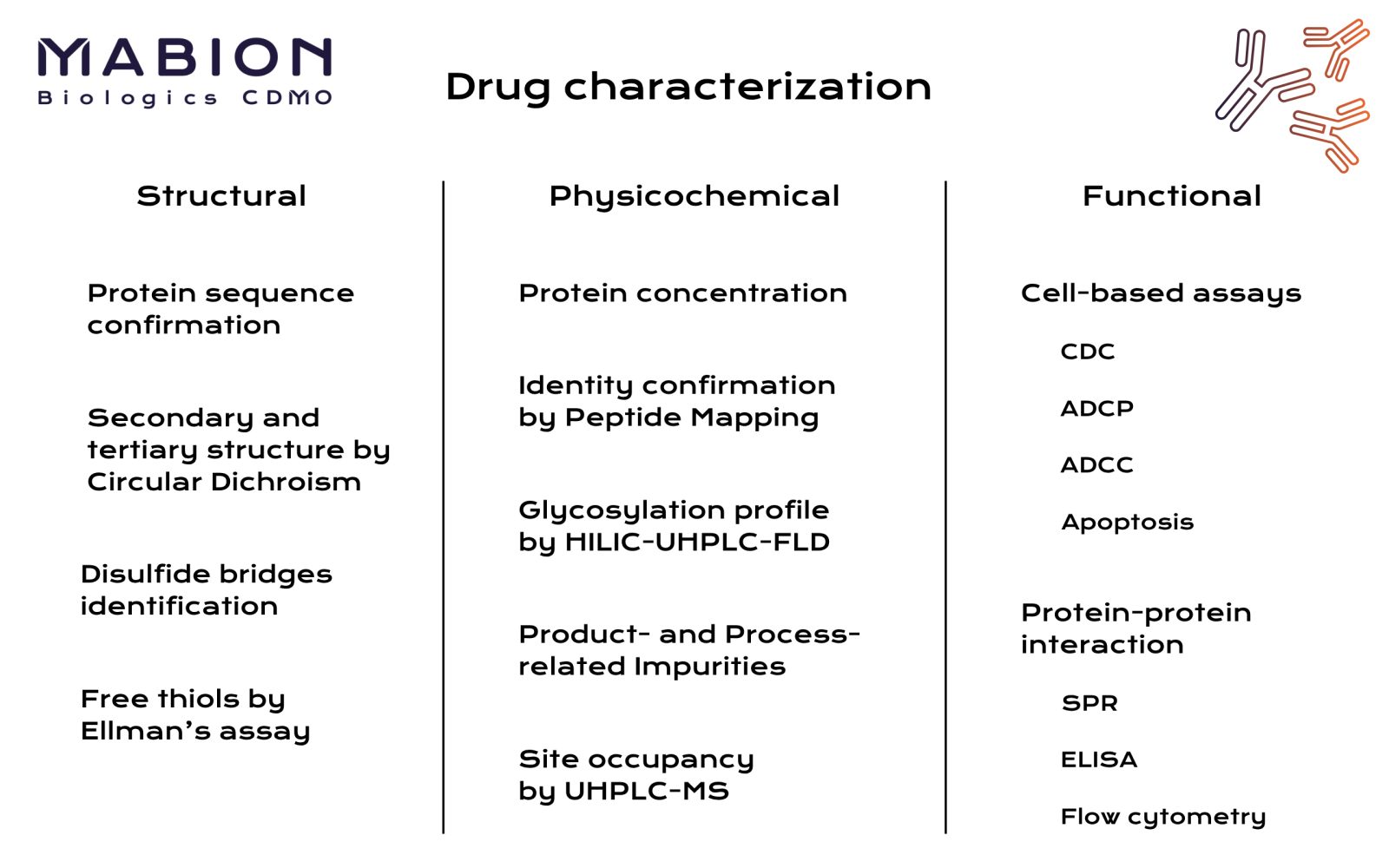
Structural characterization techniques are employed to confirm the protein’s primary structure and higher-order structure. For example, peptide mapping by liquid chromatography–mass spectrometry (LC-MS) is used to verify the amino acid sequence and identify post-translational modifications. High-resolution mass spectrometry can pinpoint modifications like deamidation or oxidation and confirm disulfide bond arrangements. Chromatographic methods (e.g. reversed-phase and ion-exchange HPLC) and capillary electrophoresis are routinely used to separate and analyze size and charge variants of the biologic. These physicochemical techniques reveal product heterogeneity such as aggregates, fragments, or charge isoforms and quantify purity. They adhere to guidelines like ICH Q6B, which outlines that structural biologics characterization should include analyses of sequence, terminal residues, disulfide bridges, glycosylation, and other modifications.
Functional characterization methods are equally important for biologics. Since many biologics (like monoclonal antibodies) act by binding targets or stimulating receptors, assays are used to measure biological activity. Binding assays such as enzyme-linked immunosorbent assays (ELISA) and surface plasmon resonance (SPR) determine the antibody’s affinity and specificity for its antigen. SPR provides kinetic data (on-rate/off-rate) and active concentration, complementing ELISA for a more complete immunological profile. Cell-based bioassays are performed to measure functions like antibody-dependent cell-mediated cytotoxicity (ADCC) or cytokine neutralization, which reflect the biologic’s mechanism of action. For example, a cytotoxicity assay can confirm that an antibody triggers immune effector cells as expected. These functional tests ensure the biologic’s potency meets required levels and that any changes in structure (such as glycosylation differences) have not diminished its intended activity.
Multiple advanced analytical techniques are often used in combination to achieve a comprehensive characterization of biologics. Spectroscopic methods like circular dichroism (CD) and Fourier-transform infrared (FTIR) spectroscopy probe secondary and tertiary structure, confirming the protein’s correct folding. Differential scanning calorimetry (DSC) assesses thermal stability and can compare higher-order structure among batches. Newer methods such as hydrogen–deuterium exchange mass spectrometry (HDX-MS) and nuclear magnetic resonance (NMR) have emerged to characterize higher-order structure and dynamics in solution.3
Meanwhile, innovations like the multi-attribute method (MAM) use high-resolution LC-MS with automated data analysis to simultaneously monitor multiple product quality attributes. This approach can replace several conventional assays by identifying and quantifying multiple site-specific modifications in one workflow. In practice, a full characterization program will leverage a “toolbox” of orthogonal methods – chromatographic, electrophoretic, spectroscopic, and bioassay techniques – to build a complete picture of a biologic’s identity, purity, and activity.4
Biologics Characterization for Ensuring Product Quality
Analytical characterization is fundamentally about ensuring that a biologic drug’s quality attributes align with those required for safety and efficacy. Each biologic has a set of critical quality attributes (CQAs) – properties like the precise molecular structure, glycosylation pattern, potency, purity, and impurity profile – that must be maintained within defined ranges to guarantee product quality. Through extensive biologics characterization, developers identify these CQAs and establish control strategies around them. For example, antibody-drug conjugates (ADCs) are characterized by their drug-to-antibody ratio (DAR), which strongly influences their efficacy and safety; one of the most important analytical tasks is to accurately determine DAR and ensure it stays consistent. If DAR drifts too high or low, the ADC’s potency or toxicity could change. By using methods like mass spectrometry and hydrophobic interaction chromatography to measure DAR, manufacturers maintain this key attribute within specifications, directly ensuring consistent product performance.2
Thorough biologics characterization also helps maintain purity and impurity control, which is crucial for biologic quality. Biologics often display various product-related variants (e.g. deamidated forms, oxidized forms, fragments) and process-related impurities (such as host cell proteins or DNA). Analytical tools can detect and quantify these at very low levels, allowing manufacturers to refine purification processes and set acceptance criteria. For instance, high-molecular-weight aggregate species are closely monitored by size-exclusion chromatography or light scattering techniques, because aggregates have been linked to increased immunogenicity in patients.5
Biologics characterization also ensures potency and consistency of clinical effect by tying structural attributes to biological function. If characterization reveals a change in a molecule’s structure (for example, a lower level of terminal sialic acids on an antibody’s glycans or a different charge variant distribution), scientists can evaluate whether this correlates with any drift in bioactivity. Often, small structural differences do not impact clinical performance – and regulators will accept quantitative changes in a CQA as long as safety and efficacy are uncompromised. Glycosylation variant might slightly alter an antibody’s Fc receptor binding, but if potency assays and clinical data show no meaningful difference, such a change is deemed acceptable. Characterization provides the evidence for these determinations. In practice, deep analytical knowledge of structure–function relationships enables a quality by design approach: manufacturing processes are designed and adjusted to consistently hit the target ranges of key quality attributes associated with optimal efficacy.6
By rigorously characterizing attributes, Mabion ensures that each lot of product released meets the predefined quality standards that correlate with expected clinical performance. In sum, comprehensive characterization underpins product quality by confirming that a biologic’s critical attributes are exactly as they should be for a safe and effective therapy
How Biologics Characterization Supports Consistency Across Batches?
Due to the inherent variability of biological production systems, demonstrating batch-to-batch consistency is a central goal of biologics characterization. Regulatory authorities and manufacturers recognize that it is neither practical nor necessary to make every batch of a biologic molecule absolutely identical at the molecular level. Instead, the emphasis is on ensuring each batch falls within a tightly controlled range of analytical attributes such that all batches are highly similar to one another. Consistency is achieved through rigorous in-process controls and thorough characterization of each lot. The best a manufacturer can do with a complex biologic is to demonstrate manufacturing consistency, with product attributes remaining within acceptable specifications for every lot. When analytical testing shows that critical quality attributes of new batches match those of prior batches (or the reference standard), we gain confidence that patients will experience the same safety and efficacy from batch to batch.7
Analytical comparability assessments are particularly vital whenever changes are made in the manufacturing process or when scaling up production. Changes might include a new cell line or facility, modified purification steps, or equipment upgrades. According to regulatory guideline ICH Q5E, the manufacturer must demonstrate that the post-change product is comparable to the pre-change product in terms of quality, safety, and efficacy. This is accomplished by an array of sensitive analytical tests applied to pre- and post-change material in a head-to-head study. If the analytical results show the two products are highly similar (within the normal variability of the methods and reference product), then it can often be concluded that no significant differences in clinical performance will occur. In fact, an underlying principle of comparability is that under certain conditions, protein products may be considered comparable on the basis of analytical testing alone. For instance, when we introduce a process improvement at Mabion, we extensively characterize multiple batches made before and after the change. This includes side-by-side comparisons of primary structure by LC-MS, higher-order structure by spectroscopy, impurity profiles, and bioassays. Modern analytical techniques are extremely powerful, so that any potential differences between batches will be uncovered. In many cases, demonstrating analytic comparability (with no new adverse impurities or loss of activity detected) is sufficient to confirm product consistency without extensive clinical studies.8
Real-world manufacturing experience has shown that robust analytical characterization can ensure consistency across decades of production. A notable example is the monoclonal antibody infliximab, which has been produced in multiple sites over more than 150 million vials while maintaining a highly consistent quality attribute profile. This consistency was achieved by tightly controlling the process and continuously monitoring the product’s analytical attributes. Deep product knowledge and diligent analysis meant that any drift in attributes was promptly detected and corrected.8 Even minor changes in raw materials or process parameters could lead to subtle drift in glycosylation or charge variants over time. However, with a rigorous analytical program in place, such changes are either prevented by design or caught within the allowable range (evolution) and shown to have no impact on safety or efficacy. Manufacturers and regulators work together to define what level of variability is acceptable for each attribute, based on clinical relevance. As long as each batch’s characterization data fall within those limits, the product is considered analytically comparable and thus clinically consistent.6
Regulatory Expectations for Biologics Characterization
Regulatory agencies require an extensive characterization data package for biologics at every stage – from initial license application to post-approval changes – to ensure product quality and consistency. Guidelines such as ICH Q6B explicitly outline the tests and acceptance criteria needed to characterize biotechnological products. This includes thorough determination of physicochemical properties (molecular identity, primary and higher-order structure, heterogeneity) and biological activity, as well as analysis of purity and impurities. Authorities expect that manufacturers use state-of-the-art analytical techniques to characterize the product as fully as possible.9 At 2016 European Medicines Agency’s monoclonal antibody guideline10 and the US FDA’s guidance emphasize detailed structural characterization (sequence confirmation, glycan profiling, variant analysis) and functional assays demonstrating the mechanism of action.
When a biologic manufacturing process is modified or during the manufacturing of biosimilars, regulators apply the principle of comparability or biosimilarity, which heavily relies on analytics. The FDA and EMA endorse a stepwise approach for biosimilars that “relies heavily on analytical methods” to demonstrate a high degree of similarity to the reference product. The goal is to show that the proposed biosimilar and the original biologic have no clinically meaningful differences, and this conclusion is driven first and foremost by comparative characterization data. In fact, advancements in analytical science have enabled regulators to reduce the requirements for large clinical trials in some cases, because sensitive analytics can now confirm products are highly similar at the molecular level.11
Regulatory agencies also require ongoing characterization and quality monitoring post-approval. Companies must establish reference standards and employ stability-indicating assays to monitor the product over its shelf life. For example, periodic re-characterization of retained samples might be requested to confirm that product quality has not drifted over years of production. Any changes in manufacturing (raw materials, equipment, scale) must be notified or approved with supporting comparability data. Health authorities expect that manufacturers have a robust analytical control strategy in place – meaning critical quality attributes are routinely tested and controlled with suitable methods. The experience over decades is that originator biologics often undergo numerous manufacturing changes (process optimizations, site transfers) after approval, and regulators have approved these based on strong comparability packages showing product consistency. They have set a precedent that if you can prove analytically that a post-change product is “highly similar” to the pre-change product, and all critical attributes remain in range, clinical re-testing may not be needed.8
Challenges and Emerging Trends in Biologics Characterization
As biologics become more complex and analytical technology advances, the field of biologics characterization is evolving rapidly. One major challenge is the sheer complexity and heterogeneity of modern biopharmaceuticals. Next-generation modalities like bispecific and multispecific antibodies, fusion proteins, and heavily glycosylated molecules present new analytical hurdles. For instance, bispecific antibodies with asymmetric architectures can mispair their polypeptide chains during expression – a problem requiring innovative analytical solutions. Advances in protein engineering have created remedies to encourage correct pairing, but they in turn drive the demand for more sensitive tools to detect any mispaired species.
In response, researchers have developed orthogonal methods: one example is a three-pronged approach for asymmetric bispecifics using intact-mass LC-MS for rapid screening, specialized HIC chromatography for quantitative lot release, and two-dimensional LC-MS for definitive peak identification. Such multi-method toolboxes are now necessary to fully characterize complex formats. Similarly, antibody–drug conjugates add another layer of complexity with a distribution of drug loads; techniques like mass spectrometry, capillary electrophoresis, and UV-vis spectroscopy must be combined to accurately profile the DAR and conjugation sites. The heterogeneity inherent in these advanced biologics means that a broader range of analytical modalities is needed than ever before.12
On the horizon, new analytical technologies are continually being introduced, and they are rapidly becoming part of the characterization toolkit. One example is the multi-attribute method (MAM), which uses peptide mapping by LC-MS not only to confirm sequence but to quantitatively monitor multiple post-translational modifications simultaneously. Many companies are adopting MAM in quality control laboratories, as it has the potential to streamline or even replace several conventional assays by consolidating them into one high-resolution MS assay.4
Furthermore, as regulatory frameworks become more accepting of innovation, there is interest in applying these advanced analytics not just to new biologics but also to streamline biosimilar development. Over two decades of experience have shown that comprehensive analytic similarity assessments can often substitute for lengthy Phase III Clinical Trials. Looking ahead, stakeholders are discussing even further streamlining: for instance, with decades of accumulated analytical knowledge, one can envision a future where routine efficacy trials for biosimilars are eliminated entirely in favor of analytic and pharmacokinetic evidence.11
Conclusion
Biologics characterization is the scientific backbone of ensuring therapeutic quality, safety, and consistency across development and manufacturing. Mabion Biologics CDMO integrates analytical methods spanning structural, functional, and comparability assessments to create a unified understanding of each product’s critical quality attributes. This holistic analytical approach enables the company to identify even subtle molecular variations, safeguarding product integrity throughout the lifecycle. By maintaining tight alignment between upstream, downstream, and analytical disciplines, Mabion guarantees that every biologic entering clinical trials is fully characterized, stable, and compliant with regulatory standards.
Through continuous innovation in mass spectrometry, bioassay design, and advanced multi-attribute methods, Mabion transforms biologics characterization into a predictive and preventive science. The company’s one-site model ensures rapid data integration and decision-making, improving both quality assurance and production agility. This integration strengthens confidence in batch-to-batch reproducibility, allowing customers to rely on consistent, high-performance biologics ready for clinical and commercial success.
FAQ
Prepared by:

Jakub Knurek
Marketing Specialist
References
- Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014; 28(4): 363-372.
- Wagh A, Song H, Zeng M, Tao L, Das TK. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. MAbs. 2018; 10(2): 222-243.
- Alhazmi HA, Albratty M. Analytical Techniques for the Characterization and Quantification of Monoclonal Antibodies. Pharmaceuticals (Basel). 2023; 16(2): 291.
- Rogers RS, Nightlinger NS, Livingston B, Campbell P, Bailey R, Balland A. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. MAbs. 2015; 7(5): 881-890.
- Fernandez-Mendivil C, Kinsella NM, Ebbers HC. A Retrospective Analysis of the Potential Impact of Differences in Aggregates on Clinical Immunogenicity of Biosimilars and their Reference Products. Clin Pharmacol Ther. 2024; 115(5): 1122-1131.
- Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011; 29(4): 310-312.
- Melsheimer R, Calmann M, DeRitis A, Philip V, Van Gog F, Doolittle L, Goyal K, Neblock D. Ensuring Product Quality, Consistency and Patient Supply over Time for a Large-Volume Biologic: Experience with Remicade®. BioDrugs. 2018; 32(5): 405-414.
- Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004; 22(11): 1383-1391.
- International Conference on Harmonisation (ICH). Q6B. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. 1999.
- European Medicines Agency. Development, production, characterisation and specifications for monoclonal antibodies and related products. 2016.
- Tuszyner A. The New Era of Biosimilar Development: Seizing the Opportunity Under EMA’s Streamlined Guidelines. Mabion Science Hub. 2025.
- Wang C, Vemulapalli B, Cao M, Gadre D, Wang J, Hunter A, Wang X, Liu D. A systematic approach for analysis and characterization of mispairing in bispecific antibodies with asymmetric architecture. MAbs. 2018; 10(8): 1226-1235.
- Christl LA, Woodcock J, Kozlowski S. Biosimilars: The US Regulatory Framework. Annu Rev Med. 2017; 68: 243-254.
Related resources
Monoclonal Antibody Prophylaxis for Infectious Diseases – Passive Immunization and Prevention
Monoclonal antibody, Vaccines

End-to-End Manufacturing of Biosimilars – From Cell Line to Commercial Product
Biosimilars, Manufacturing

Microbiological Cleaning in Cleanrooms in CDMO Operation
GMP, Microbiology