Development and regulation of veterinary monoclonals
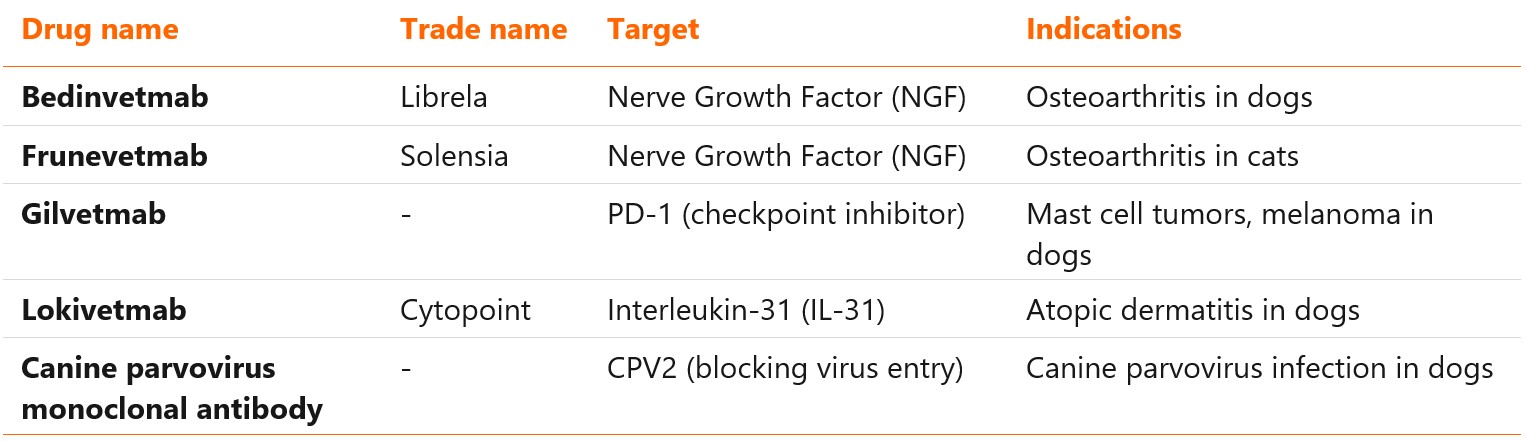
- Veterinary monoclonal antibodies are a relatively new class of therapeutics, lagging 30 years behind the human mAbs. Only five molecules have been approved so far: bedinvetmab, frunevetmab, lokivetmab, gilvetmab and canine parvovirus monoclonal antibody (the last two with conditional approval).
- Simarly to the drugs for human use, veterinary drugs must pass a strict registration procedure, both in the EU and US realms – sponsoring companies must present comprehensive evidence that they are safe and effective for use. While in the EU veterinary mAbs are approved by the EMA, in the U.S. these products are evaluated either by FDA or USDA (U.S. Department of Agriculture), depending on the intended indication.
- Clinical development of veterinary mAbs is more streamlined that for human monoclonals. Typical pathway includes laboratory studies, which confirm the effectiveness on appropriate animal model, followed by field studies with client-owned animals.
- Many innovative mAbs are currently being investigated and pharmaceutical companies count the time until first veterinary biosimilars can be introduced to market. However, many years must pass before the patents for veterinary products expire and so far no specific regulations have been issued by drug agencies.
In contrast to the monoclonal antibodies for human use, veterinary mAbs are a much less recognized class of therapeutics, even among the bio-tech professionals. Yet it is a rapidly developing field, with many new molecules approved in the recent years, and even more in development. Despite the high costs, veterinary mAbs have achieved quite a substantial commercial success, mostly because they are indicated for popular companion animals and target their most common diseases (such as osteoarthritis or atopic dermatitis).
Approved veterinary mAbs
Three veterinary monoclonal antibodies have been authorized so far by regulatory authorities in Europe and United States1,2:
- Bedinvetmab (Librela)
- Frunevetmab (Solensia)
- Lokivetmab (Cytopoint)
Another two, glinvetmab and canine parvovirus monoclonal antibody, received conditional approval in United States in 20233,4. Lokivetmab, authorized in December 2016, was the first veterinary mAb to reach the market5. Targets, types and approved indications of these antibodies are listed in the table below.
Both in the EU and the US, regulatory approval of veterinary medicines is governed by drug agencies, which also handle the drugs for human use (in case of the U.S., the approval of certain mAbs is covered by another agency, which is exclusively dedicated to veterinary drugs, see below). The procedural steps and scope of information that must be provided by the pharmaceutical company bear many similarities to human medicines6.
Regulatory approval in European Union
In the EU, the decision to approve or not approve a veterinary drug is issued by a dedicated body called Committee for Veterinary Medicinal Products (CVMP), which is analogous to the Committee for Human Medicinal Products (CHMP) responsible for the authorization of human drugs5. The approval process is addressed by a separate regulation, namely the “REGULATION (EU) 2019/6 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 11 December 2018 on veterinary medicinal products and repealing Directive 2001/82/EC”7. The first guidance on monoclonal antibodies for use in animals has been issued by EMA in 20178.
Similarly to the human products, applications for veterinary drugs derived from biotechnological processes must be submitted via a centralized procedure. Companies that wish to obtain approval must include a comprehensive data package (so called ‘dossier’), which proves that the medicine in question meets the quality, efficacy and safety standards defined by the EU regulations and guidelines. The authorization procedure is essentially the same as for human drugs. The evaluation by EMA takes up to 210 days, which can be interrupted to allow applicants to prepare responses and supply additional data on agency’s request. The final decision is made by CVMP and the official marketing authorization is granted by the European Commission. The manufacturers are also encouraged to obtain regulatory and scientific advice throughout the development process6.
Medicinal products for food-producing animals must undergo extra evaluation, including consumer safety6. However, so far, none of the registered mAbs are authorized for use in livestock.
Regulatory approval in United States
The situation is more complicated in the United States. The regulatory pathway for veterinary monoclonal antibodies depends on their intended indication. Among the currently available antibodies, bedinvetmab and frunevetmab (both used for the alleviation of pain associated with osteoarthritis) were approved by the FDA, while lokivetmab, used in atopic dermatitis, as well as Canine Parvovirus mAb, were approved by USDA9.
At the FDA, all veterinary products must go through the New Animal Drug Application (NADA) procedure and the division responsible for licensing veterinary products is Center for Veterinary Medicine (CVM)10. As for human medicines, the Sponsor must collect comprehensive information about the safety and effectiveness of the developed drug. This is done by conducting the studies described in the submitted Investigational New Animal Drug (INAD) – a product-specific file, which outlines the development program plan for the candidate molecule. The Sponsor then submits a NADA to CVM, which includes the studies’ results and also the proposed label. In the end, the team of experts at CVM reviews the submitted application and issues the final decision10.
Apart from the USDA pathway, the US regulation of veterinary medicines is analogous to the one existing for human products. The main difference lies in the names of the respective offices and documents – CVM instead of CDER, INAD instead of IND and NADA in place of NDA. Biologic and non-biologic drugs are proceeded by the FDA in the same manner.
Development of veterinary antibodies
Clinical development of veterinary drugs is less complicated and less risky than for human drugs. Researchers can confirm efficacy at an early stage by running laboratory studies with the use of established animal model of disease (if available). Such studies require only a small number of individuals and allow for close and objective monitoring. The initial findings must be later confirmed in full-scale field studies, but the probability of failure is much reduced by the initial investigations. Field trials typically enroll several hundred participants, with clinical evaluations conducted in a blind manner by both veterinarians and pet owners. Researchers must ensure the enrolment of representative populations e.g., the sufficient number of dog races.
Clinical development of veterinary mAb is well pictured by the clinical trials of lokivetmab11. Laboratory investigations consisted of PK evaluation, proof-of-concept and dose-determination studies. Proof-of-concept study was conducted to obtain preliminary evidence of efficacy in a IL-31 induced canine model of pruritus. The same model was also used to determine the appropriate clinical dose. Lokivetmab was observed to attenuate the adverse effects of IL-31 challenge, showing that IL-31 inhibition is a promising strategy for treating atopic dermatitis in dogs11.
Laboratory studies were then followed by field trials with client-owned dogs: a pilot study with 78 dogs and two large-scale studies performed in the US and EU, which enrolled 211 and 274 participants, respectively. The US trial used a placebo control, while the EU trial compared lokivetmab to active, standard-of-care control (cyclosporine, immunosuppressant widely employed in AD treatment). Both provided overwhelming evidence of efficacy, leading to the prompt authorization by regulatory agencies. For example, the treatment success rate after 4 weeks in the US trial was 69% on high-dose lokivetmab, compared to 29% on placebo11. The drug has gained much popularity, contributing to the excellent financial results of the manufacturing company, Zoetis (total revenue of over $2 bln in the third quarter of 2023)12.
Many new veterinary monoclonal antibodies are being developed at this moment. One of them is ranevetmab, which is another anti-NGF antibody planned to be used in osteoarthritis13. Zoetis, a developer of frunevetmab, dislosed recently that the company is working on tens of new antibodies for allergies, dermatological conditions, cardiac disorders and cancers14. As veterinary mAbs were approved only recently and the regulatory pathway for veterinary biosimilars is not yet clear, the potential biosimilar products are not going to appear any soon. However, the discussion in subject literature has already begun and several companies are likely to initiate the clinical development in the near future15.
Although veterinary monoclonals lag 30 years behind the human products, next decades will certainly witness the approval of many new breakthrough mAbs for treating both companion and farm animals.
FAQ
Prepared by:

Regulatory Compliance Specialist
References
1. Paul Ehrlich Institute (PEI). Medicinal products: Immunological Veterinary Medicines. Link: https://www.pei.de/EN/medicinal-products/veterinary/veterinary-node.html.
2. Y! Sports “Dr. Maro: Monoclonal antibodies/biologicals for pets: New medications for old problems” (June 12, 2023). Link: https://sports.yahoo.com/dr-maro-monoclonal-antibodies-biologicals-093117895.html.
3. Merck press release. “Merck Animal Health Announces Availability of Novel Canine Oncology Therapy to Veterinary Specialists Practicing Oncology Nationwide” (October 13, 2023). Link: https://www.merck-animal-health.com/blog/2023/10/13/merck-animal-health-announces-availability-of-novel-canine-oncology-therapy-to-board-certified-veterinary-oncologists-nationwide/.
4. Elanco Animal Health Inc. press release. “Elanco Announces Breakthrough Treatment for Deadly Canine Parvovirus” (May 2, 2023). Link: https://www.elanco.com/en-us/news/elanco-announces-breakthrough-treatment-for-deadly-canine-parvovirus.
5. Zoetis press release. “Zoetis receives USDA license for CytopointTM” (December 21, 2016). Link: https://www.zoetisus.com/news-and-media/zoetis-receives-usda-license-for-cytopoint.
6. European Medicines Agency (EMA). “Applying for EU marketing authorization for medicinal products for veterinary use” (2015).
7. “REGULATION (EU) 2019/6 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 11 December 2018 on veterinary medicinal products and repealing Directive 2001/82/EC”. Official Journal of the European Union (January 7, 2019).
8. European Medicines Agency (EMA). “Questions and answers on monoclonal antibodies for veterinary use” (2017). Link: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-and-answers-monoclonal-antibodies-veterinary-use_en.pdf.
9. U.S. Department of Agriculture (USDA). Licensed Veterinary Biological Product Information. Link: https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/veterinary-biologics/ct_vb_licensed_products.
10. Food and Drug Administration (FDA). “From an idea to the marketplace: The journey of an animal drug through the approval process.” (2020). Link: https://www.fda.gov/animal-veterinary/animal-health-literacy/idea-marketplace-journey-animal-drug-through-approval-process.
11. European Medicines Agency. Cytopoint European Public Assessment Report (EPAR). (February 16, 2017).
12. Zoetis. “Zoetis Announces Third Quarter 2023 Results” (November 2, 2023). Link: https://investor.zoetis.com/news/news-details/2023/Zoetis-Announces-Third-Quarter-2023-Results/default.aspx.
13. Epstein, Mark E. “Anti-nerve growth factor monoclonal antibody: a prospective new therapy for canine and feline osteoarthritis.” The Veterinary Record 184.1 (2019): 20.
14. AVMA News. “Monoclonal antibodies show promise as new therapy for veterinary patients” (November 21, 2023). Link: https://www.avma.org/news/monoclonal-antibodies-show-promise-new-therapy-veterinary-patients.
15. S Álvarez Marchante. “Regulatory viability of biosimilars in veterinary medicine” (2018). Link: https://ddd.uab.cat/pub/tfg/2018/195092/TFG_salvarezmarchante_poster.pdf.
If you are interested in our services regarding the development and regulatory aspects of monoclonal antibodies, please don’t hesitate to contact us. Mabion’s specialist offer individualized approach tailored to the biologic and clinical characteristics of the developed drug. Our experience with end-to-end development and manufacturing of biologics gives us an overwhelming advantage over other CDMOs as we perfectly understand the client’s perspective. Contact us to obtain more information on our CDMO services.