Similar but not the same: an in-depth look at the differences between EMA and FDA
Biologics, EMA, FDA, Regulatory

- Pharmaceutical professionals not involved in regulatory affairs may sometimes perceive EMA and FDA as near-mirror images, regulating medicines in different territories yet operating in a similar way. While there are many similarities between the two agencies, several significant differences do exist and can be of paramount value when preparing the drug development strategy.
- The contrasts between EMA and FDA are most evident in their legal frameworks, industry consultations and drug approval pathways. Conversely, regulations concerning clinical research, manufacturing and quality, and post-marketing surveillance are highly similar.
- Despite some critical differences, outcomes of EMA and FDA assessments are highly congruent for both biologic and non-biologic medicines. Cooperation between the two organizations is evolving, making the drug registration process more streamlined and less expensive for the pharmaceutical industry.
All medicines, including biologics, have to pass a stringent registration procedure to ensure that only effective and safe products are marketed. Regulatory agencies around the world set precise criteria that have to be met by every medicinal product in order to receive approval.1 The European Medicines Agency (EMA) and Food and Drug Administration are the two most renown agencies that govern drug approval procedures in the European Union and United States, respectively.2,3 They are often viewed as counterparts, functioning in essentially the same way and differing mostly with respect to the covered region. However, despite many similarities and widespread efforts to standardize regulatory processes, such as adoption of ICH guidelines and joint scientific meetings, many important differences do exist, creating numerous challenges to pharmaceutical industry. Companies need to prepare separate strategies, hire additional personnel and sometimes conduct additional studies to satisfy requirements of both agencies.
In this article we review some of the key dissimilarities between EMA and FDA regulatory approach, with special attention to biologic drug development (both innovative and biosimilar).
Legal framework
The most basic difference between the EU and US agencies lies in their legal background and jurisdiction. The FDA is responsible for regulating a wide range of products for human and animal use in the US, including medicines, medical devices, tobacco products, cosmetics and most foods.4 In contrast, the EMA governs only human and veterinary medicines and participates in the process of medical devices evaluation.5 After Brexit, the EMA’s jurisdiction covers European Union, Norway, Iceland and Liechtenstein.6 The United Kingdom and Switzerland have their own regulatory bodies – Medicines and Healthcare Products Regulatory Agency (MHRA) and Swiss Agency for Therapeutic Products (Swissmedic).7,8 Both organizations cooperate closely with the EMA, exchanging vital information and harmonizing the procedures.9,10
Whereas the FDA has a direct authority to approve the drug products, EMA can only evaluate submissions and provide non-binding recommendations to the European Commission (EC).11,12 The committees responsible for this process are Committee for Medicinal Products for Human Use (CHMP) and Committee for Medicinal Products for Veterinary Use (CVMP).13,14 In theory, the EC may not concur with the CHMP/CVMP’s assessment and issue a negative final decision. However, in practice this never happens.
Clinical research
In both EU and US settings, clinical research is tightly regulated and conducted in accordance with the Good Clinical Practice (GCP) guidelines in order to protect the rights, health and well-being of participants.15 To initiate a clinical study, sponsors are required to obtain permission from authorities. In the US, these permissions are of course granted by the FDA – pharmaceutical companies or investigators must submit the Investigational New Drug (IND) application, which contains data from pre-clinical studies, information on manufacturing process and clinical development plan for a candidate drug.16 The IND is reviewed by the FDA and if no safety concerns are raised within 30 days after the submission, the sponsor can initiate the clinical trials. In the EU, the procedure is less centralized and despite the fact that only a single application is submitted via a common electronic platform (Clinical Trial Information System, CTIS), national authorities in each of the member countries must issue separate permissions.17 In both regions, approval from regulatory bodies must be followed by an approval from Institutional Review Boards (IRBs) or Ethics Committees (ECs).18 All initiated trials are required to be registered in publicly available databases – ClinicalTrials.gov in the US and EudraCT in the EU.20,21
All over the world, medicines pass through the same clinical trial phases – initial Phase 1 first-in human studies, Phase 2 studies with preliminary efficacy/safety exploration and dose selection, Phase 3 efficacy and safety confirmatory studies, and post-approval Phase 4 trials to extend indications and/or collect further evidence on drug’s utility.22 Sometimes Phase 0 trials are also distinguished, which employ very small drug doses and involve even less subjects than typical Phase 1 studies.
Consultations
Both agencies offer scientific consultations throughout the entire drug development process and prior to dossier filing. Such consultations are carried out at EMA in the form of Scientific Advice or protocol assistance, the latter being offered exclusively to orphan drug developers.24 The goal is to obtain advice on the evidence that needs to be generated in order to prove the efficacy and safety of the developed product for the purpose of regulatory approval. Sponsors compile their questions along with the proposed solutions and background information on the product in the so-called briefing document. The response is typically received in a written form, but sometimes it can be supplemented by a face-to-face meeting with EMA representatives. Importantly, the guidance obtained from the Agency is not binding on any of the sides, and approach may change based on the later developments.
Consultations with the FDA are held in the form of official meetings. Different types of meetings are organized depending on the drug development stage and class. Separate sets of meetings are available for innovative prescription drugs, generics and biosimilars (see the below table).25,26 Recommendations issued by the FDA during the scientific consultations are regarded as more binding, though the agency may reconsider its opinion once the new important data becomes available. This is especially the case for pre-IND meetings, during which the preliminary plans for clinical development are discussed.27 Importantly, in both EMA and FDA, noncompliance with the obtained guidance can result in failed registration process.
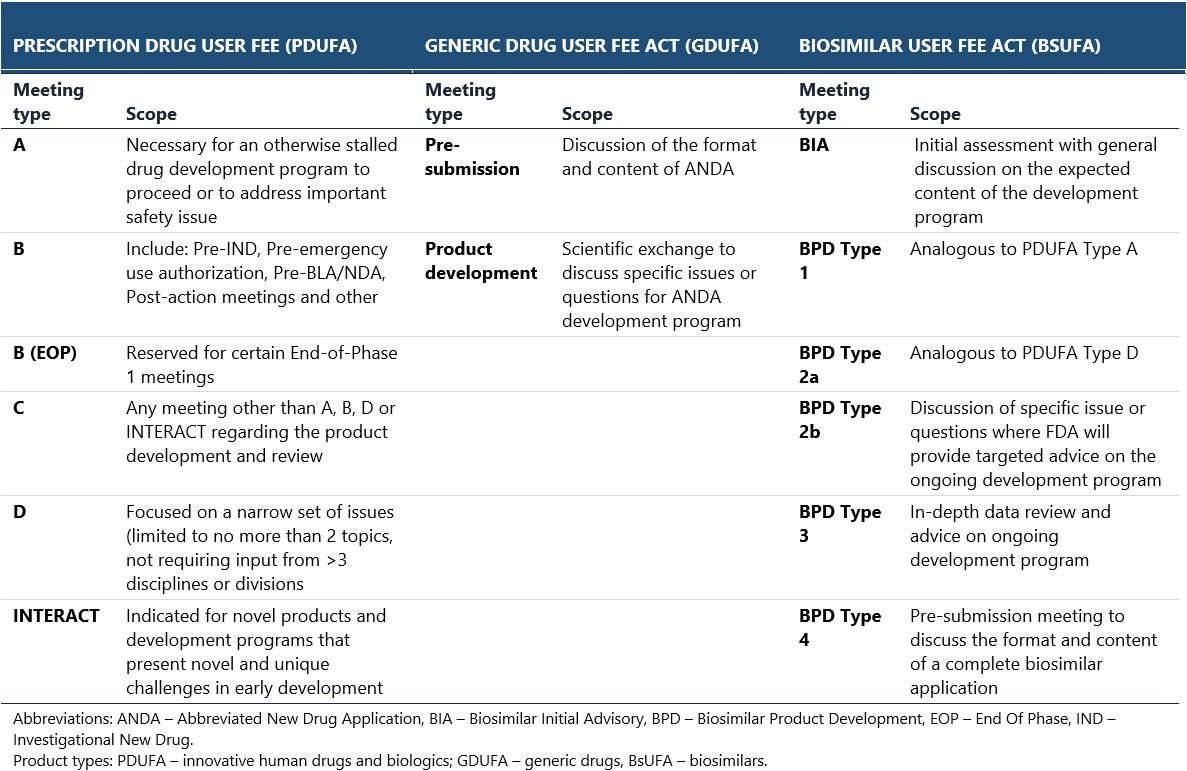
The EMA and FDA may sometimes present disparate views on particular issues that are extremely difficult to reconcile when planning a drug development program. For example, they may ask to conduct a biosimilar trial in two completely different patient populations, which cannot be enrolled within a single trial. Differences in regulatory requirements may also impede drug development: for instance, EMA has diverging requirements for drug safety evaluation in vulnerable patient populations, whereas the FDA puts more emphasis on clinical endpoints and quality-of-life indices, regardless of the population.28 In such cases, additional studies are necessary, increasing the overall financial burden and sometimes causing the expected revenues to fall below the break-even point.
To make things easier for the industry, several years ago EMA and FDA decided to create an option of parallel scientific advice (PSA), during which assessors from both agencies can concurrently exchange their views on scientific issues with the sponsors.24 PSAs are particularly useful when the developed medicinal products lack clear guidelines or there are significant differences in the EMA’s and FDA’s thinking. However there are certain limitations as to when PSA can take place and not all sponsor’s requests are positively evaluated – for a variety of reasons, one or both agencies can refuse to participate in such procedure.
Manufacturing and quality issues
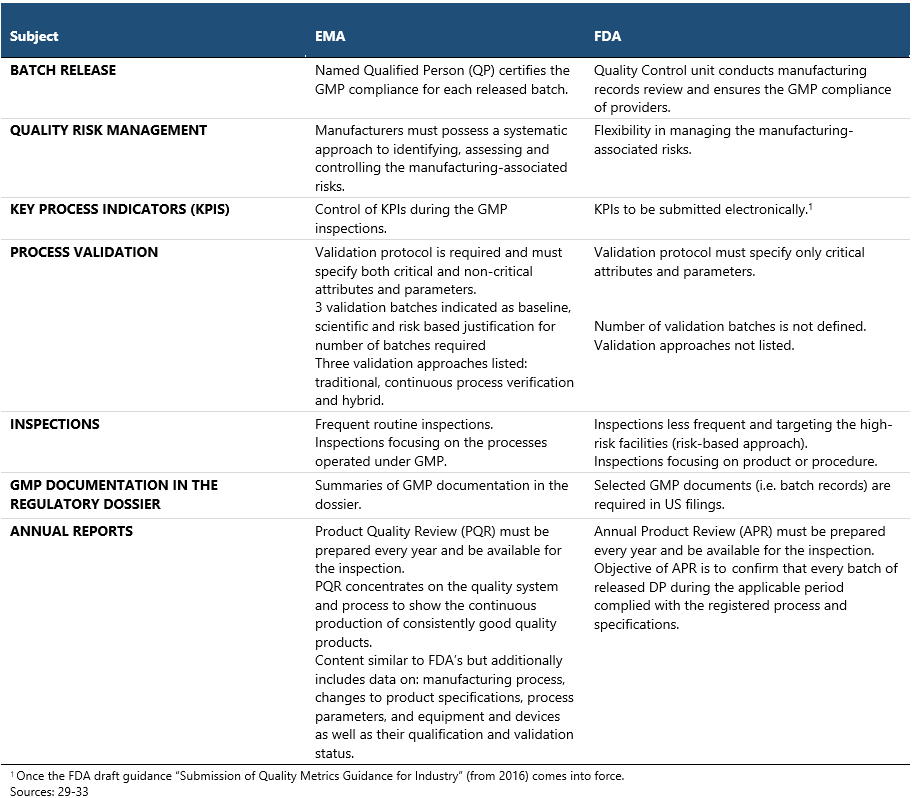
Good Manufacturing Practices (GMPs) are of paramount importance to drug regulators all over the world.29,30 They set minimum requirements for manufacturing and quality control methods that pharmaceutical companies must comply with, in order to ensure that the produced medicines can be safely administered to patients. Despite the universal adherence to the GMP principles, each authority releases its own guidelines which may present differing views on certain CMC aspects. Importantly, the FDA refers to the GMP guidelines as cGMP (c stands for “current”) to emphasize that manufacturers meet GMP using the most modern and up-to-date methods and that the regulations are constantly evolving, in line with the developments in the industry.
Some of the most important differences between EMA and FDA in the GMP approach are displayed in the table above. In general, the EU GMPs are recognized as more comprehensive and less flexible in most respects, featuring a higher volume of requirements compared to the US standards, such as the need for QP or defined number of validation batches.31 The FDA GMP inspections are regarded as more stringent though, due to the inspectors being federal employees and their swift enforcement action possibilities, such as issuing warning letters, import alerts or initiating product recalls. FDA inspections typically involve extensive review of documentation and rigorous employee interview techniques.
Manufacturers for both the EU and US markets must carefully verify the compliance with two different regulatory systems.
Approval process
In contrast to the aforementioned areas, the process of drug authorization in the EU is vastly different from the one in the US, although the structure of the submitted dossier follows the same, eCTD format.34–37
In the FDA, there are two principal approval pathways for prescription drugs: New Drug Application (NDA) for small molecules and Biologic License Application (BLA) for biologics and biosimilars.38 Biologic drugs, as defined in Section 351(1) of the PHS Act, include: viruses, therapeutic serums, toxins, antitoxins, vaccine, blood, blood components or derivatives, allergenic products, proteins (except any chemically synthesized polypeptides), or analogous product, or arsphenamine or derivative of arsphenamine (or any other trivalent organic arsenic compound).39 The goal of both applications is essentially the same, to obtain marketing authorization for the product by providing sufficiently robust evidence that it is safe and effective to be used in sought indication(s). Nevertheless, there are slight differences when it comes to the application content and review process.
In both pathways, the applicants prove that the developed manufacturing process is capable of providing a drug product of adequate potency, quality and purity. However, this task is much more challenging for biologic drugs, which are complex molecules produced in living cells.41 For example, the purity of drug must be backed by more extensive data showing that no extraneous material of biologic nature was introduced during the drug production. The more sophisticated (and at the same time more risky) manufacturing process makes the pre-approval inspection from FDA inevitable. Such inspections are not always carried out in case of NDA, where FDA utilizes rather a risk-based approach.41
While the dossiers for small molecules are reviewed by Center for Drug Evaluation and Research (CDER), BLAs are directed to either CDER or another FDA’s division called Center for Biologics Evaluation and Research (CBER) depending on the type of biologic drug. CDER oversees monoclonal antibodies and other therapeutic proteins, immunomodulating drugs and growth factors.42,43
In the European Union, there are four regulatory pathways for drug approval36:
- Centralized
It is obligatory for all medical products derived from biotechnological processes, advanced therapy products, orphan drugs and novel medicines indicated for the treatment of cancer, diabetes, HIV/AIDS as well as neurodegenerative, viral and autoimmune diseases.
- Mutual recognition
It is applicable solely to the medicinal products already authorized in at least one EU member state.
- Decentralized
It can be used for all medicines for which the centralized procedure is not required. It allows to register the product in more than one EU member countries in parallel. Decentralized procedure is the most commonly used approval pathway.
- National
As the name suggests it is used by Sponsors who wish to market a drug product in only one member state. Such procedure may be later followed by the mutual recognition pathway. Marketing authorization is issued by a national regulatory agency.
As previously mentioned in the Legal framework section, in contrast to FDA, EMA has no authority to issue the drug approvals. Instead, the CHMP committee, consisting of representatives from each EU country, reviews the submitted dossier and provides recommendation to the European Commission, which is then responsible for making the final decision.13 The CHMP handles both biologic and non-biologic medicines.
There are additional dissimilarities between EMA and FDA that are worth mentioning here:
- FDA requests the submission of raw data from clinical studies and performs their own analysis during the application review. EMA has no such policy in place and relies entirely on the data processed by the Applicant. However, this may soon change as in 2022 the EU agency announced the pilot project on raw data analysis.44,45
- FDA uses advisory committees and panels, consisting of medical professionals, patient representatives and other concerned parties, which support the agency in decision-making process. They comment on the submitted applications and vote for or against the drug approval. However, the voting results are not binding for the FDA. EMA has no such committees in place.
- There are some additional approval categories specific to the US, endowing the sponsor with special marketing rights. For example, biosimilars may be granted an interchangeability status which means that pharmacies can exchange the reference product for biosimilar without provider’s permission. In Europe, originators can be substituted for generics or biosimilars and there is no need for introducing a special regulatory category.
While historically EMA and FDA agree in their drug assessments almost 100% of the time, there have been some notable exceptions. Most disagreements stem from divergent interpretations of scientific evidence though some touched upon the regulatory issues as well. The most spectacular historic examples of the differences in EMA and FDA assessments are weight loss medications lorcaserin (Belviq) and phentermine/topiramate (Qsymia).46 Clinical trials of both drugs have pointed to the potential increase in the risk of cardiovascular events. Although the results in each case failed to reach statistical significance, similar experience with previously marketed but now withdrawn weight loss medications (fenfluramine and dexfenfluramine) should have acted as a clear warning sign that the observed safety signal is real. The EMA took more cautious approach and refused marketing authorization on the ground of increased cardiovascular risk that cannot be excluded with the available data. In case of Belviq, the CHMP cited a concern over drug-induced valvulopathy (serious disease affecting valves of the heart); for Qsymia there was a potentially elevated risk of stroke and heart attack, caused by an increased heart rate shortly after the drug administration. The FDA reached more positive conclusion on the risk-benefit balance of both products, and ignoring aforementioned uncertainties associated with cardiovascular safety profile, gave them a green light. However, at the same time it requested their manufacturers to conduct additional post-marketing trials that would confirm or exclude the potentially increased risk of heart attacks, strokes and other cardiac events. In the hindsight, the EMA position proved to be wrong – subsequent studies determined that lorcaserin and phentermine/topiramate are not associated with heart attacks or strokes (although lorcaserin was later withdrawn due to unrelated safety issues).47,48 Nevertheless, the FDA received sharp criticism for basing the approval on incomplete data and not considering the grave implications had the risk been confirmed.
Another, less serious case, involves abaloparatide – an osteoporosis drug initially approved by the FDA but not by the EMA.49 The chief reason for refusing marketing authorization was the lack of statistically significant reduction in nonvertebral fractures following the exclusion of two GCP-incompliant sites. Efficacy against this endpoint is spelled out in EU guidelines, whereas no such requirement exists in US regulations.50
Biosimilars regulations
The introduction of biosimilars was a real game-changer from the public health perspective, decreasing the huge costs of biologic treatment and making them more accessible to patients of lower socioeconomic status. Now, both EU and US citizens benefit from the availability of tens of such products, including the most successful monoclonal antibodies used in the treatment of autoimmune and oncologic conditions (e.g. rituximab, trastuzumab, adalimumab). However, the development of biosimilars market and regulatory environment in these regions has followed different paths.
The EMA introduced biosimilars guidelines many years ahead of the FDA, resulting in an early market growth and fierce competition between numerous biosimilar brands. The first biosimilar in the EU was licensed in 2006 (Omnitrope, containing somatotropin).51,52 In contrast, the first US biosimilar, Zarxio (containing filgrastim) was approved nine years later in 2015.51,53 Initially, the level of similarity testing and the time required for the FDA to approve a new product, significantly exceeded those of the EMA. However, since that time the registration process has been successfully streamlined and now the FDA approves many biosimilars several months before the European agency, overtaking EMA’s position as a leader in this sector.54 In some cases the approval is granted by the FDA without late-phase clinical studies (filgrastim/pegfilgrastim biosimilars including Nivestym® and Udenyca®). This resulted in rapid increase in biosimilar’s uptake and substantial decrease in healthcare costs.55
The EMA and FDA share similar views regarding the biosimilars development.56 Both define biosimilars in essentially the same way as a biological medicine that is highly similar to the already approved one, for which there are no clinically meaningful differences to the reference in terms of safety, quality and efficacy.57,58 However, companies are bound to use as a reference product a biological medicine originating from the given territory. Thus, the EMA application must be based on studies performed with the EU-sourced reference, while for FDA application it is necessary to use the US-sourced product.
In terms of biosimilars development, the Quality Target Product Profile (QTPP) must be based on reference product and requires its strict monitoring over time. In contrast, innovative drugs allow the manufacturers for more flexibility during development. Understanding the nuances between biosimilars and innovative drugs development and their impact on project timelines is critical.
To streamline the biosimilar development for both markets, pharmaceutical companies frequently perform the so-called bridging between the EU and US originators.59 In the biosimilars universe, bridging typically consists of a direct physicochemical comparison of the 3 products (biosimilar and two references) and a 3-way PK similarity study performed to show equivalence in their pharmacokinetic profiles.60 Three comparisons are run in order to determine equivalence: Reference A and Reference B, Biosimilar and Reference A, and Biosimilar and Reference B. A study success can be claimed only when all comparisons are met. Successful bridging allows to extrapolate the results of a pivotal efficacy trial performed with one reference product to cover another reference product e.g., data for the EU originator are treated as if they were generated using US originator. This cost-effective strategy obviates the need for large, expensive Phase III trials using both references and is particularly useful when a company seeks to enter another market after completing registration in one region.
The evidence required for biosimilar approval is generally consistent across the EU and US. Both jurisdictions apply similar standards of pharmaceutical quality, safety and efficacy to both innovative biologics and biosimilars.61 The goal of the development program is not to prove once again that the contained active substance in safe and effective for a given indication, which would require running a lengthy and costly trials, but establish similarity to the originator at all mentioned levels. However, unlike innovative biologics, where clinical trial data are paramount, the hierarchy of evidence for biosimilars places greatest emphasis on CMC data having the greatest influence on regulatory decisions.61
Post-marketing surveillance
Once a medicinal product is granted with approval, it becomes subject to post-marketing safety surveillance, comprising passive and active safety monitoring.62
Passive surveillance relies on public reporting of adverse events by patients, physicians or other involved persons. Marketing authorization holders in both the EU and US, are mandated to collect the data on adverse events suspected to be caused by their drug. Regulatory authorities also collect adverse event data and analyze them for potential safety signals. Unfortunately, the clinical trials frequently lack the statistical power to detect rare but serious adverse reactions – they can only be discovered by observing a larger patient cohort using the drug outside the research setting.63,64 Furthermore, clinical trials are typically conducted on narrowly defined patient groups which do not represent the much broader population using the drug after authorization.65
EMA and FDA utilize separate but similar AE monitoring tools: EudraVigilance and FAERS, respectively.66,67 Additionally, in the US there is a dedicated database for reporting vaccine-related AEs called VAERS.68 Anyone can submit a report to these systems by filling out special forms in an electronic or paper format.
Active surveillance consists of post-authorization safety studies conducted by manufacturers or regulatory agencies.69,70 If there are lingering safety concerns from the drug approval process, both EMA and FDA can request additional studies focusing on potential adverse events. Typically such studies are of observational nature, though randomized controlled trials may be deemed more appropriate in certain cases.
Based on the data from passive or active post-marketing surveillance, regulatory agencies may opt to withdraw the product, modify labelling to include new safety information and/or mandate additional confirmatory studies. Some differences between EMA and FDA pharmacovigilance regulations pertain to the risk management process.71 In the EU, all manufacturers are required to provide during the registration procedure a document called Risk Management Plan (RMP), which outlines the pharmacovigilance system and risk mitigation measures to be implemented by the company. The document is created regardless if serious safety concerns were identified during the review process and is based on an overall safety profile of approved molecule. The RMP can be adjusted on request of the member country to align with the local legal requirements. An analogous risk management program within the US regulatory framework is called Risk Evaluation and Mitigation Strategies (REMS). In contrast to the RMP required by EMA, REMS applies only to specific medicines for which serious safety concerns have been identified during the application review. The proposed risk mitigation strategies apply solely to these concerns and do not consider an overall safety profile of the licensed drug. REMS is uniform across all US territory and cannot vary in individual states.
Consequences of separate pathways
The existence of separate regulatory systems in the EU and US, each marked by substantial differences in procedures, requirements and expectations, presents numerous challenges for companies seeking to register their products on both markets. Guidelines and recommendations obtained from the FDA may conflict with those obtained from the EMA, sometimes making it necessary to conduct additional EU- or US-specific studies. This not only rises financial burden but also complicates the development pathway and prolongs the time-to-market period. Regulatory risk escalates as well; for instance, the additional clinical study may yield contradictory findings to the first one or prompt additional queries from the agencies. Harmonization of regulatory requirements would undoubtedly mitigate this risk and increase the number of innovative drugs reaching both markets. This would translate into huge benefits for the pharmaceutical industry, especially the small to medium enterprises that lack the resources to run multiple trials.
Current and future collaboration
While the full unification of EU and US regulatory processes belongs to fiction, the cooperation between EMA and FDA is steadily growing, progressing towards greater data sharing, joint advisory procedures and harmonization of guidelines.72
Over the recent decades, EMA and FDA have entered into several important collaborations aimed at expediting regulatory decisions, accelerating drug development and increasing patients’ safety. Throughout their regulatory and scientific processes, both agencies routinely exchange confidential data from pre-clinical and clinical studies, post-marketing surveillance (adverse drug reaction reports) and GCP/GMP inspections. They also consult each other during the parallel scientific advice procedure (see Consultations section).
However, the most significant milestone in the history of EMA-FDA cooperation is the signing of the mutual recognition agreement (MRA) in 2017, which became fully operational in 2019.73 This agreement enables both agencies to rely on each other’s GCP/GMP inspections, share information on inspections and quality defects, and waive batch testing of products on import into their territories.
In addition to data sharing and joint meetings, EMA and FDA regularly hold virtual meetings in the so-called “clusters” – areas of mutual interest to both agencies that require closer collaboration and intensified exchange of information.72 These clusters encompass diverse domains such as advanced therapy medicinal products (ATMPs), biosimilars, blood products, oncology-hematology medicines, orphan drugs, pediatric drugs, pharmacogenomics and pharmacovigilance.
Hopes for parallel assessment procedures were sparked in 2011, when EMA and FDA launched Quality by Design Pilot Program aimed at ensuring the consistent implementation of ICH Q8, Q9 and Q10 guidelines, and harmonizing regulatory decisions made by the two agencies.74 The program was run for over five years with the final report published in 2017. In total, 14 applications were evaluated but despite the highly positive feedback expressed by both regulators, no further steps were taken. However, cooperation between EMA and FDA is poised to increase over time. Recently, both organizations announced the expansion of parallel scientific advice procedure to cover complex generics/”hybrid medicines”.75 Complex generics are a growing group of drug products that possess complex active ingredients, formulation or administration routes or that constitute a medicine-device combination. Their development is particularly complicated for generic drug companies, limiting the availability of more affordable products for patients. Additional significant initiatives are anticipated in the coming years.
Prepared by:

Adam Tuszyner
Registration Dossier Specialist, Pharmacovigilance Specialist

Anna Małecka
Regulatory Manager
References
- Van Norman, Gail A. “Drugs and devices: comparison of European and US approval processes.” JACC: Basic to Translational Science 1.5 (2016): 399-412.
- European Medicines Agency (EMA) homepage. Link: https://www.ema.europa.eu/en/homepage.
- Food & Drug Administration homepage. Link: https://www.fda.gov/.
- Food & Drug Administration “What does FDA regulate?”. Link: https://www.fda.gov/about-fda/what-we-do/what-does-fda-regulate.
- European Union. “European Medicines Agency”. Link: https://european-union.europa.eu/institutions-law-budget/institutions-and-bodies/search-all-eu-institutions-and-bodies/european-medicines-agency-ema_en
- European Medicines Agency (EMA), “Marketing authorization”. Link: https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation.
- Medicines and Healthcare Products Regulatory Agency (MHRA) homepage. Link: https://www.gov.uk/government/organisations/medicines-and-healthcare-products-regulatory-agency.
- SwissMedic homepage. Link: https://www.swissmedic.ch/swissmedic/en/home.html.
- ECA Academy, “MHRA announces Cooperation with seven international Partners.” (June 7, 2023). Link: https://www.gmp-compliance.org/gmp-news/mhra-announces-cooperation-with-seven-international-partners.
- European Medicines Agency (EMA), “Bilateral interactions with non-US regulators”. Link: https://www.ema.europa.eu/en/partners-networks/international-activities/bilateral-interactions-non-eu-regulators.
- European Medicines Agency (EMA), “Authorization of medicines”. Link: https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines.
- Food & Drug Administration (FDA), “Development & Approval Process”. Link: https://www.fda.gov/drugs/development-approval-process-drugs.
- European Medicines Agency (EMA), “Committee for Medicinal Products for Human Use (CHMP)”. Link: https://www.ema.europa.eu/en/committees/committee-medicinal-products-human-use-chmp.
- European Medicines Agency (EMA), “Committee for Veterinary Medicinal Products (CVMP)”. Link: https://www.ema.europa.eu/en/committees/committee-veterinary-medicinal-products-cvmp.
- ICH Harmonized Guideline: Integrated Addendum To Ich E6(R1): Guideline For Good Clinical Practice E6(R2), Step 4 version (Nov 9, 2016).
- Food & Drug Administration (FDA) “Investigational New Drug (IND) Application” (July 20, 2022). Link: https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application.
- European Medicines Agency (EMA), “Clinical Trials Information System (CTIS)”. Link: https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/clinical-trials-human-medicines/clinical-trials-information-system.
- Mackintosh, Douglas R., and Vernette Molloy. “Differences in clinical trial conduct in US and EU investigational sites.” The Quality Assurance Journal: The Quality Assurance Journal for Pharmaceutical, Health and Environmental Professionals 5.1 (2001): 13-17.
- Gherghescu, Ioana, and M. Begoña Delgado-Charro. “The biosimilar landscape: an overview of regulatory approvals by the EMA and FDA.” Pharmaceutics 13.1 (2020): 48.
- https://clinicaltrials.gov/.
- https://eudract.ema.europa.eu/.
- Gad, Shayne Cox, ed. Clinical trials handbook. John Wiley & Sons, 2009.
- Reference 3.
- European Medicines Agency (EMA), “Scientific advice and protocol assistance”. Link: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/scientific-advice-protocol-assistance.
- Food & Drug Administration (FDA), Guidance document: Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products (Sep 2023). Link: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/formal-meetings-between-fda-and-sponsors-or-applicants-pdufa-products.
- Food & Drug Administration (FDA), Formal Meetings for CBER-regulated products (Jan 2024). Link: https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber/formal-meetings-cber-regulated-products.
- National Institute of Health (NIH) Clinical Center America’s Research Hospital, FDA Responses and Meetings for Investigational New Drug Applications. Link: https://www.cc.nih.gov/orcs/ind4.html.
- Image Analysis Group “Getting approval: the EMA and the FDA.” Link: https://www.ia-grp.com/views/regulatory-approval-fda-ema/.
- European Medicines Agency (EMA). Good manufacturing practice. Link: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compliance-research-development/good-manufacturing-practice.
- Food & Drug Administration. Current Good Manufacturing Practice (cGMP) Regulations. Link: https://www.fda.gov/drugs/pharmaceutical-quality-resources/current-good-manufacturing-practice-cgmp-regulations.
- ECA QP Association, “What are the differences between EU and FDA GMP?” (01 Aug 2018). Link: https://www.qp-association.eu/qpag_news_6550.html.
- Schmitt, Wolfgang “FDA and EU GMP – always in balance?“ GMP Journal (15 May 2018).
- BioBoston Consulting “EU vs US GMPs: Key differences.” (29 Jun 2023) Link: https://www.linkedin.com/pulse/eu-vs-us-gmps-key-differences-biobostonconsulting/.
- European Medicines Agency (EMA). Guidance for Industry on Providing Regulatory Information in Electronic Format: Harmonized Technical Guidance for eCTD Submissions in the EU. Version 4.0 (2016).
- Food & Drug Administration (FDA). Electronic Common Technical Document (eCTD) (22 Mar 2023). Link: https://www.fda.gov/drugs/electronic-regulatory-submission-and-review/electronic-common-technical-document-ectd.
- European Medicines Agency (EMA). Authorization of medicines. Link: https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines.
- Food & Drug Administration (FDA). Development and Approval Process (08 Aug 2022). Link: https://www.fda.gov/drugs/development-approval-process-drugs.
- Food & Drug Administration (FDA). Types of Applications (23 Oct 2014). Link: https://www.fda.gov/drugs/how-drugs-are-developed-and-approved/types-applications.
- Food & Drug Administration (FDA). Public & Health Service Act. Link: https://www.govinfo.gov/content/pkg/COMPS-8773/pdf/COMPS-8773.pdf.
- Food & Drug Administration (FDA). New Drug Application (NDA). Link: https://www.fda.gov/drugs/types-applications/new-drug-application-nda.
- Allucent. “What are regulatory differences between NDA and BLA?”. Link: https://www.allucent.com/resources/blog/what-are-regulatory-differences-between-nda-and-bla.
- Food & Drug Administration (FDA). Center for Biologics Evaluation and Research (CBER). Link: https://www.fda.gov/about-fda/fda-organization/center-biologics-evaluation-and-research-cber.
- Food & Drug Administration (FDA). Transfer of Therapeutic Biological Products to the Center for Drug Evaluation and Research. Link: https://www.fda.gov/combination-products/jurisdictional-information/transfer-therapeutic-biological-products-center-drug-evaluation-and-research.
- Food & Drug Administration (FDA). Study Data for Submission to CDER and CBER. Link: https://www.fda.gov/industry/study-data-standards-resources/study-data-submission-cder-and-cber.
- European Medicines Agency (EMA). “EMA launches pilot project on analysis of raw data from clinical trials” (12 Jul 2022). Link: https://www.ema.europa.eu/en/news/ema-launches-pilot-project-analysis-raw-data-clinical-trials.
- Wolfe, Sidney M. “When EMA and FDA decisions conflict: differences in patients or in regulation?.” BMJ 347 (2013).
- Bohula, Erin A., et al. “Cardiovascular safety of lorcaserin in overweight or obese patients.” New England Journal of Medicine 379.12 (2018): 1107-1117.
- Ritchey, Mary E., et al. “Cardiovascular safety during and after use of phentermine and topiramate.” The Journal of Clinical Endocrinology & Metabolism 104.2 (2019): 513-522.
- Eladynos: EPAR – Refusal public assessment report.
- European Medicines Agency (EMA). Guideline on the evaluation of medicinal products in the treatment of primary osteoporosis. CPMP/EWP/552/95 Rev. 2.
- Niazi, Sarfaraz K. “The coming of age of biosimilars: a personal perspective.” Biologics 2.2 (2022): 107-127.
- Omnitrope European Public Assessment Report (EPAR). Link: https://www.ema.europa.eu/en/medicines/human/EPAR/omnitrope.
- Novartis. FDA approves first biosimilar ZarxioTM (filgrastim-sndz) from Sandoz. Link: https://www.novartis.com/news/media-releases/fda-approves-first-biosimilar-zarxiotm-filgrastim-sndz-from-sandoz.
- Jung, Emily H., et al. “FDA and EMA biosimilar approvals.” Journal of general internal medicine 35 (2020): 1908-1910.
- Carl, David L., et al. “Comparison of uptake and prices of biosimilars in the US, Germany, and Switzerland.” JAMA network open 5.12 (2022): e2244670-e2244670.
- Daller, Justin. “Biosimilars: A consideration of the regulations in the United States and European union.” Regulatory Toxicology and Pharmacology 76 (2016): 199-208.
- European Medicines Agency (EMA). “Biosimilar medicines marketing authorization”. Link: https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/biosimilar-medicines-marketing-authorisation.
- Food and Drug Administration (FDA). Biological Product Definitions. Link: https://www.fda.gov/files/drugs/published/Biological-Product-Definitions.pdf.
- Pan, Zhiying, et al. “Design and Analysis of a Biosimilar Bridging Study with a Prediction Interval-Based Consistency Test.” Therapeutic Innovation & Regulatory Science 55 (2021): 765-772.
- Food and Drug Administration (FDA). FDA’s Overview of the Regulatory Guidance for the Development and Approval of Biosimilar Products in the US. Link: https://www.fda.gov/files/drugs/published/FDA%E2%80%99s-Overview-of-the-Regulatory-Guidance-for-the-Development-and-Approval-of-Biosimilar-Products-in-the-US.pdf.
- Radovan, Diana. “Biosimilar development–an overview.” Medical Writing 28 (2019): 20-27.
- Vlahović-Palčevski, Vera, and Dirk Mentzer. “Postmarketing surveillance.” Pediatric clinical pharmacology (2011): 339-351.
- Tsang, Ruth, Lindsey Colley, and Larry D. Lynd. “Inadequate statistical power to detect clinically significant differences in adverse event rates in randomized controlled trials.” Journal of clinical epidemiology 62.6 (2009): 609-616.
- A Wahab, Izyan, et al. “The detection of adverse events in randomized clinical trials: can we really say new medicines are safe?.” Current drug safety 8.2 (2013): 104-113.
- Tan, Yen Yi, et al. “Comparing clinical trial population representativeness to real-world populations: an external validity analysis encompassing 43 895 trials and 5 685 738 individuals across 989 unique drugs and 286 conditions in England.” The Lancet Healthy Longevity 3.10 (2022): e674-e689.
- EudraVigilance – European database of suspected adverse reaction reports. Link: https://www.adrreports.eu/en/index.html.
- Food & Drug Administration (FDA). Questions and Answers on FDA’s Adverse Event Reporting System (FAERS). Link: https://www.fda.gov/drugs/surveillance/questions-and-answers-fdas-adverse-event-reporting-system-faers.
- Vaccine Adverse Event Reporting System (VAERS). Link: https://vaers.hhs.gov/.
- European Medicines Agency (EMA). Post-authorization safety studies. Link: https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/post-authorisation-safety-studies-pass.
- Food & Drug Administration. Postmarketing Clinical Trials. Link: https://www.fda.gov/vaccines-blood-biologics/biologics-post-market-activities/postmarketing-clinical-trials.
- Biomapas. “EMA & FDA: What Are the Similarities & Differences in Risk Management Procedures?”. Link: https://www.biomapas.com/ema-and-fda-risk-management/.
- European Medicines Agency (EU). Bilateral interactions with non-EU regulators: United States. Link: https://www.ema.europa.eu/en/partners-networks/international-activities/bilateral-interactions-non-eu-regulators/united-states.
- European Medicines Agency (EU). Mutual recognition agreements. Link: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compliance-research-development/good-manufacturing-practice/mutual-recognition-agreements-mra.
- ECA Academy. “EMA-FDA QbD Pilot Program Report” (10 May 2017). Link: https://www.gmp-compliance.org/gmp-news/ema-fda-qbd-pilot-program-report.
- Food & Drug Administration (FDA). CDER’s OGD and EMA’s Parallel Scientific Advice Pilot Program for Complex Generics Works to Increase Harmonization and Bring Generic Drugs to Patients. Link: https://www.fda.gov/drugs/our-perspective/cders-ogd-and-emas-parallel-scientific-advice-pilot-program-complex-generics-works-increase.
- Regulatory Focus, RAPS https://www.raps.org/news-and-articles/news-articles/2021/11/fda-official-says-increasing-number-of-enforcement.
If you are interested in our services regarding the development and regulatory aspects of biologic drugs, please don’t hesitate to contact us. Mabion’s specialist offer individualized approach tailored to the biologic and clinical characteristics of the developed drug. Our experience with end-to-end development and manufacturing of biologics gives us an overwhelming advantage over other CDMOs as we perfectly understand the client’s perspective. Contact us to obtain more information on our services.
Related resources

Mass spectrometry in peptide and protein analysis
Analytics, Drug substance, Proteins

Time to Market Strategy – 5 CDMO Tactics to Speed Up Biologic Launches
Biologics, Drug development, Manufacturing

Navigating OOX Results: Effective Analysis and Management in CDMO Laboratories
Analytics, Manufacturing, Regulatory