The New Era of Biosimilar Development: Seizing the Opportunity Under EMA’s Streamlined Guidelines
Biosimilars, Clinical trials, Drug development, EMA, FDA, Mabion, Regulatory
- Major change in regulatory framework for biosimilars is coming: EMA’s reflection paper on biosimilars is calling for abandoning the costly Phase III efficacy studies with FDA heading in the same direction.
- The introduction of streamlined pathway will substantially reduce the development costs by as much as 50%, creating opportunity for small-to-medium enterprises to join the EU and US market.
- End-to-end CDMOs with multi-year experience in biosimilar development, such as Mabion, are best-positioned to help companies thrive in this new regulatory landscape.
Current bottlenecks in biosimilar development
The Biosimilar developers today face significant challenges under the traditional development paradigm. Bringing a biosimilar to market typically takes 7-9 years at a cost often exceeding $100–$200 million. A major contributor is the large Phase III confirmatory trial, which can enroll hundreds of patients and last several years. These comparative efficacy trials are extremely costly – often tens of millions of dollars (commonly representing up to ~50% of a developer’s total budget). This lengthy, expensive process not only delays patient access to cheaper biologics but also creates substantial financial risk, forming a high barrier to entry for smaller biotech companies. In summary, key bottlenecks include:
- High Phase III trial costs: A single large Phase III can cost $20–50+ million, consuming a huge portion of development funds.
- Extended time-to-market: Prolonged clinical trial timelines (often 2-3 years for Phase III) delay regulatory submission and launch.
- Funding and access hurdles: The capital and expertise needed for lengthy trials favor big pharma; smaller firms struggle to finance development, limiting competition and innovation.
These challenges have spurred industry and regulators to seek more efficient pathways that maintain rigorous safety standards while reducing unnecessary duplication of clinical testing.
EMA’s long-awaited proposal: scientific and economic rationale
Regulators are now reconsidering whether large efficacy trials are truly needed for biosimilars that are proven highly similar to their reference products. In April 2025, the European Medicines Agency (EMA) released a draft “Reflection paper on a tailored clinical approach in biosimilar development” calling for a more streamlined, case-by-case clinical program.1,2 The paper’s key message is that when a biosimilar’s structural, functional, and pharmacokinetic (PK) profiles are virtually indistinguishable from the reference drug, additional Phase III efficacy studies may not add value. Advances in analytical science and extensive positive experience with biosimilars support this shift:
- Strong analytical similarity predicts clinical performance: It is now well-documented that if a biosimilar is highly similar in chemistry and mechanism, clinical outcomes will match the reference.3 In fact, regulators note that to date no biosimilar with robust CMC/analytic similarity has ever failed to demonstrate equivalent efficacy. This track record suggests the large confirmatory trials rarely uncover new information beyond what earlier comparability studies show.
- Consistently positive efficacy results: Over 40 biosimilars have been approved in the EU, almost all with Phase III data, yet none has shown a meaningful efficacy difference.4 This supports the view that a properly conducted comparability exercise (analytics + PK/PD) is a “sensitive assay” for biosimilarity, whereas outcome trials are often insensitive and redundant.
- Notable precedents without Phase III: Regulatory agencies have already approved certain biosimilars using only PK/PD studies. For example, a filgrastim (G-CSF) biosimilar was approved by FDA without any Phase III efficacy trial, relying on Phase 1 PK/PD and immunogenicity data.4,5 Filgrastim’s clear pharmacodynamic markers (absolute neutrophil count) enabled a purely analytical and PK approach – a model now extended to other products with straightforward mechanisms and surrogates.6
EMA’s reflection paper explicitly questions the need for dedicated efficacy trials, especially for less complex biologics with well-understood mechanisms. It argues that modern analytical tools and biological assays can ensure two proteins that are “highly similar” will bind the same targets and have the same clinical effect. In such cases, efficacy and safety can be inferred from demonstrating physicochemical and functional similarity plus PK equivalence. In other words, if no meaningful structural or bioactivity differences are detected in vitro, a costly efficacy trial is unlikely to reveal any difference except random noise.
From an economic perspective, this tailored approach could be transformational. Waiving certain clinical data requirements would simplify development and evaluation while still upholding the EU’s high safety standards.1,4 By eliminating an apparently unnecessary Phase III, biosimilars could reach the market faster without compromising quality. This benefits healthcare systems (sooner access to affordable biologics) and developers (significantly lower costs and risk). Notably, the EMA is not alone in this thinking:
Filgrastim and other early examples: As noted, filgrastim set an example in which a Phase III was waived and approval was based on PK/PD equivalence. Other products with readily measurable PD markers (e.g. insulin analogues, growth hormones) have similarly been approved with minimal or no efficacy trials.12 These cases provided real-world proof that streamlined programs can still ensure safety and efficacy, prompting regulators to formally reconsider guidelines.
FDA & Health Canada alignment: Other regulators are actively discussing similar reductions. In September 2023, the FDA (with international regulators) held a workshop titled “Increasing the Efficiency of Biosimilar Development Programs – Reevaluating the Need for Comparative Clinical Efficacy Studies” acknowledging growing consensus that many biosimilar Phase III trials can be skipped.7 By early 2025, the FDA signaled openness to Phase III waivers on a case-by-case basis, and indeed the first such waivers have been granted in the US. For instance, Formycon (a biotech company) was allowed to terminate a planned Phase III for a pembrolizumab (Keytruda) biosimilar after FDA agreed it was not necessary, given strong Phase I and analytical data.8 Similarly, Sandoz announced it would minimize Phase III for its Keytruda biosimilar following FDA advice.9 Just recently, the US agency has issued an upfront waiver of clinical efficacy study for ustekinumab biosimilar developed by prof. Niazi, a well-known figure in the world of biosimilars who has persistently advocated for abandoning the Phase III trial paradigm.10 Health Canada has gone a step further by releasing a draft guidance in 2025 removing the requirement for comparative efficacy/safety trials in most biosimilar submissions.11 Under the proposed Canadian policy, a biosimilar would generally only need comparative PK data to establish equivalence, plus continued monitoring of safety and immunogenicity. This global regulatory convergence underscores the robust scientific rationale for a leaner approach.
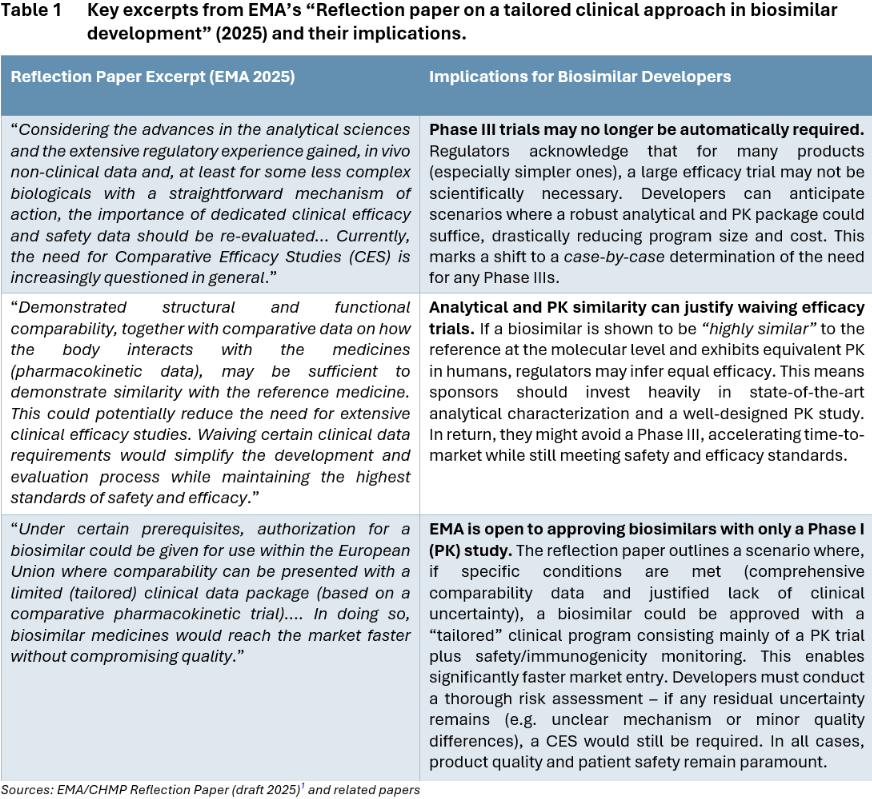
Overall, the EMA’s reflection paper and parallel discussions by FDA and Health Canada represent a paradigm shift. Regulators are essentially saying: If you can prove your biosimilar matches the reference product in every scientifically relevant way (structure, function, PK, etc.), we might not require you to prove it again in an expensive efficacy trial. The public consultation on EMA’s draft runs until September 30, 2025, but it reflects a clear trend that is already taking hold via case-by-case waivers.1 Developers should prepare for a new normal where the emphasis of clinical development shifts to smaller, targeted studies rather than large Phase III. Table 1 above highlights several key statements from the EMA’s draft reflection paper (2025) and discusses their practical meaning for biosimilar developers.
Impact on biotech business if Phase III is waived
The prospect of a Phase III waiver isn’t just a regulatory curiosity – it could fundamentally improve the economics of biosimilar development. By slashing the most expensive and time-consuming part of the development program, companies can reduce costs, accelerate timelines, and lower the bar for entry. Some estimated impacts include:
- Cost Reduction: Eliminating Phase III (and related interchangeability studies) could cut total development costs by up to $150 million, from the current staggering amount of ~$300-325 million.13 Avoiding a large trial saves not only direct trial expenses but also indirect costs (drug supply, monitoring, overhead) and frees resources to develop more products in parallel.
- Faster Time-to-Market: Without Phase III and interchaengability studies, the overall development timeline could shrink by 2–3 years. Instead of ~8 years, a biosimilar might reach approval in ~5-6 years.13 This accelerated timeline means earlier entry into the market, which in turn can yield a significant competitive advantage (the first biosimilar to market often secures a larger market share before additional competitors arrive).
- New Emphasis on Phase I and PD Markers: In a streamlined pathway, the comparative PK study becomes the pivotal trial. Developers will put greater emphasis on designing efficient Phase I studies that may also include pharmacodynamic (PD) endpoints when available. Sensitive PD biomarkers (e.g. receptor occupancy, cytokine levels, etc.) that reflect the drug’s mechanism can strengthen the similarity argument and potentially serve in lieu of efficacy endpoints.2 The goal is to extract maximum information about biosimilar performance from a small number of healthy volunteers or patient samples.
- Case-by-Case Immunogenicity/Safety Evaluation: Immunogenicity will remain a key consideration, but sponsors might address it with smaller, focused studies instead of broad Phase III monitoring. Regulators like Health Canada propose that safety and immunogenicity data be gathered within the comparative PK study or in dedicated short-term studies. In practice, this means developers must still conduct thorough immunogenicity assays (e.g. anti-drug antibody testing) and comparative safety assessments, but these could be done in a more targeted way. The reflection paper notes that a Phase I trial alone, with its smaller size, may not fully characterize rare adverse events2; therefore, post-marketing surveillance or risk management plans could gain importance when Phase III is waived.
- Reliance on CMC and Advanced Analytics: Perhaps the most significant impact is the increased importance of a robust CMC (Chemistry, Manufacturing and Controls) package and analytical comparability. If clinical testing is pared back, regulators will scrutinize the quality and similarity data even more. Developers must invest in state-of-the-art analytical methods (e.g. high-resolution mass spectrometry, functional bioassays, characterization of post-translational modifications) to prove “no clinically meaningful differences” from the reference. As one regulator put it, the revised approach “emphasiz[es] the need for extensive comparative quality studies to demonstrate a high degree of similarity”. Companies will need top-tier analytical and process development capabilities to meet this standard, including lot-to-lot consistency data, stability studies, and possibly novel in vitro assays that can substitute for clinical endpoints. In short, analytical rigor will make or break biosimilar applications in the new paradigm.
From a business standpoint, these changes can lower the financial barrier and encourage more players – including smaller biotechs – to enter the biosimilar arena. Reduced development expense improves the potential return on investment, which is crucial as biosimilar profit margins are narrower than those of novel biologics. Moreover, earlier market entry means more revenue before pricing pressures fully kick in. However, companies must also adapt by bolstering their early-stage development capabilities: success will depend on excellence in analytics, clever study design, and regulatory savvy rather than large clinical operations.
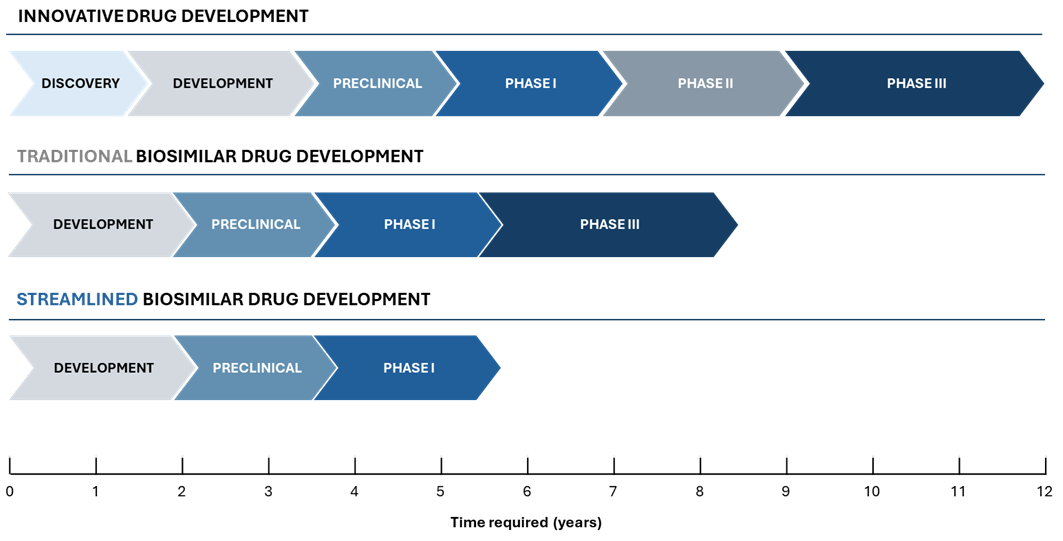
Streamlined versus traditional development timeline
The conventional route includes one or more Phase I PK/PD studies (e.g. preliminary research in healthy volunteers and PK briding between the EU and US products) and a Phase III comparative efficacy study (CES), extending the timeline to 7-9 years before approval. The streamlined approach omits the Phase III trial, relying on analytical characterization and a comparative PK/PD study for a total timeline of ~5-6 years. This CES waiver path accelerates market entry by focusing on critical similarity data early.
As illustrated above (Figure 1), the opportunity to waive Phase III can significantly compress the development program. In the traditional pathway, after completing laboratory analytics and Phase 1, a sponsor would spend several years executing Phase 3 trials in a large patient population to confirm efficacy and safety before filing for approval. In the streamlined pathway, once robust analytical similarity is established and Phase 1 PK/PD comparability is demonstrated, the sponsor can proceed directly to regulatory submission. This eliminates lengthy trials, allowing the biosimilar to reach patients years sooner. Importantly, the overall requirements for demonstrating biosimilarity do not lessen – they are simply addressed with more efficient tools (analytical and PK data) rather than redundant efficacy trials. Developers pursuing the tailored route must ensure that by the end of the comparability exercise, all residual uncertainties are addressed through analytics or targeted studies. If successful, the reward is a faster, leaner development cycle that can be the difference between being the first to market versus the third or fourth entrant.
How Mabion can support developers in the new landscape
For biosimilar companies looking to capitalize on this industry shift, partnering with an experienced CDMO is invaluable. Mabion, as a European CDMO with a unique biosimilar heritage, is well positioned to guide developers through a streamlined approach.
By partnering with Mabion, biosimilar developers can confidently pursue the new streamlined pathway without compromising on quality.
Our end-to-end capabilities and expertise help ensure that even without a Phase III, nothing is left to chance in demonstrating similarity. In particular, Mabion offers:
- Analytical Development & Similarity Assessment: We have cutting-edge analytical laboratories to perform extensive comparability studies. This includes physicochemical characterization (structural analyses, glycosylation profiling, impurity profiling) and biological functional assays. Our scientists design studies to meet the latest EMA and ICH guidelines, generating the data needed to justify a Phase III waiver. We emphasize “state-of-the-art, orthogonal methods” as per the reflection paper’s expectations5, giving regulators confidence in the biosimilar’s quality.
- Cell Line and Process Development: Mabion can develop high-expression cell lines and robust manufacturing processes for your biosimilar, ensuring consistent quality from lab scale through commercial scale. A robust process is essential because product consistency underpins the comparability exercise. Our team’s deep experience with monoclonal antibodies and other biologics (including our in-house rituximab biosimilar program) means we understand the critical quality attributes that must be controlled. We also offer process optimization to fine-tune attributes – for example, adjusting glycosylation patterns – early, so that your biosimilar meets similarity targets and avoids comparability pitfalls.
- GMP Manufacturing for Clinical and Commercial Supply: As a one-stop CDMO, Mabion provides Good Manufacturing Practice (GMP) production of both clinical trial material and commercial batches. We can manufacture material for Phase 1 PK/PD studies quickly and then seamlessly scale up for launch, which is crucial if development timelines are compressed. Our facilities equipped with 2,500L Cytiva bioreactors meet the EU and FDA standards, having passed numerous GMP inspections from various regulatory authorities. We have relevant experience with products for highly regulated markets, which means fewer delays and reliable supply as you accelerate toward approval.
- Regulatory Strategy & Consultation Services: Navigating the tailored approach requires strategic regulatory planning. Mabion’s regulatory affairs experts help map out a development strategy aligned with EMA’s reflection paper and other agencies’ latest thinking. We conduct gap analyses of your data package against regulatory expectations and identify if any supplemental evaluations (e.g. an immunogenicity substudy or PD endpoints) may still be needed. We also support clients in seeking scientific advice meetings with agencies to obtain a Phase III waiver approach. Our team has direct experience interacting with EMA, FDA, and other authorities on biosimilar requirements, which can greatly de-risk the regulatory process.
- Project Management & Cost/Time Modeling: With leaner development comes the need to carefully manage parallel workstreams. Mabion offers program management to coordinate analytical, non-clinical, and clinical tasks efficiently. We use detailed cost and timeline modeling tools to compare scenarios (traditional vs streamlined) and optimize your development plan. By simulating different pathways, we can help demonstrate the significant savings of a tailored approach and ensure stakeholders (investors, partners) are on board.
Overall, Mabion provides a fully integrated solution, from clone development to commercialization, specifically tailored for biosimilars. Uniquely, we draw on our own experience as a biosimilar developer: for example, Mabion’s work on a rituximab biosimilar (MabionCD20) has honed our expertise in comparability exercises and immunogenicity assessment. By partnering with us, biosimilar sponsors can confidently pursue the new streamlined paradigm, knowing that no aspect of quality or compliance will be overlooked. Our mission is to enable clients to bring affordable biotherapeutics to patients faster and more cost-effectively, without compromising on excellence.
Conclusion
The biosimilar industry is entering a new era where “bigger” development is not always better – smarter development is. Regulators across Europe and North America are embracing science-based, streamlined development models that eliminate redundant clinical trials. This marks a collective shift towards efficiency in biosimilar approval, a win-win for manufacturers, healthcare systems, and patients. By reducing development burdens, these changes lower the hurdles for bringing biosimilars to market, allowing even smaller biotech players to compete and innovate.
Lean development should not be confused with easy development – it still requires excellence in CMC, analytics and regulatory strategy, which can be achieved by working with experienced CDMO.
Early evidence of this shift is already visible. For instance, Dr. Reddy’s Laboratories, an emerging-market company, recently launched its first biosimilar in the highly regulated UK market – Versavo (bevacizumab) – which was originally developed and approved in India.14 In 2024 they also managed to register in the EU a biosimilar to rituximab (Ituxredi).15 This example shows that with the right capabilities, smaller local companies can succeed globally in the biosimilar space. As development pathways streamline, we expect to see more such companies seizing opportunities, not just the traditional big pharma firms. In fact, over the next decade, the pool of opportunities is enormous: reference biologics totaling an estimated $200 billion in value will lose exclusivity by 2030. The companies that can navigate the new leaner development process will be poised to capture these markets by being first or fast followers with high-quality biosimilars. In this dynamic landscape, speed and efficiency of development translate directly to competitive advantage. Every month shaved off the timeline can mean millions in additional revenue and broader patient reach. Conversely, an unnecessary Phase III trial could be the difference between a commercially viable product and a missed opportunity. The industry consensus is clear – if a biosimilar’s analytical and PK data make its similarity evident, forcing a large efficacy trial is no longer justified.1 The focus is shifting to “get it right” in the lab and early clinics, rather than “prove it again” in Phase III.
Mabion is excited about this paradigm shift because it aligns with what we have long believed – that a rigorous scientific approach can ensure biosimilar safety and efficacy more efficiently than the old routines. We have positioned our services to help clients bring out the best from this wave of change. Whether you are a small biotech aspiring to develop your first biosimilar, or a seasoned generics company looking to expand your biologics portfolio, embracing a tailored clinical development approach could be the key to success. Lean development does not mean easy development – it requires excellence in CMC, analytics, and regulatory strategy. With Mabion’s support as an experienced CDMO partner, you can confidently reduce unnecessary steps and focus resources where they matter most.
In conclusion, the industry is moving toward an era of faster, smarter biosimilar development driven by regulatory innovation. Companies that adapt early will not only cut costs and timelines, but also improve their chances of capturing market share by launching at the earliest possible date. Patients worldwide stand to benefit as high-quality biosimilars reach the market sooner, expanding access to life-saving biologics. Mabion welcomes you to contact our Business Development team to discuss how we can jointly design a development program optimized for this new reality. Together, let’s capitalize on the momentum toward leaner biosimilar development – and bring the next generation of affordable biologics to patients, faster.
Prepared by:

Adam Tuszyner
Regulatory Compliance Specialist
References
- Streamlining development and assessment of biosimilar medicines | European Medicines Agency (EMA). Link: https://www.ema.europa.eu/en/news/streamlining-development-assessment-biosimilar-medicines.
- European Medicines Agency (EMA). “Reflection paper on a tailored clinical approach in biosimilar development”. https://www.ema.europa.eu/en/documents/other/reflection-paper-tailored-clinical-approach-biosimilar-development_en.pdf.
- Frapaise, Francois-Xavier. “The end of phase 3 clinical trials in biosimilars development?.” BioDrugs 32.4 (2018): 319-324.
- Niazi, Sarfaraz K. “A timely recommendation to the european medicines agency and the FDA on rationalizing comparative clinical efficacy testing of biosimilars.” Discover Medicine 1.1 (2024): 87.
- Jung, Emily H., et al. “FDA and EMA biosimilar approvals.” Journal of general internal medicine 35.6 (2020): 1908-1910.
- Biosimilars Review and Report. (n.d.). Product profile of Pfizer’s Filgrastim Biosimilar (Nivestym). https://biosimilarsrr.com/product-profile-nevistym-2-2/.
- “Increasing the Efficiency of Biosimilar Development Programs – Reevaluating the Need for Comparative Clinical Efficacy Studies.” U.S. Food And Drug Administration, 16 July 2024, www.fda.gov/drugs/news-events-human-drugs/increasing-efficiency-biosimilar-development-programs-reevaluating-need-comparative-clinical.
- “Formycon Announces Decision on Phase III Trial With FYB206 and Provides Update on Potential Need to Adjust the Valuation of FYB202 and FYB201 – Formycon AG.” Formycon AG, 8 July 2025, www.formycon.com/en/blog/press-release/formycon-announces-decision-on-phase-iii-trial-with-fyb206-and-provides-update-on-potential-need-to-adjust-the-valuation-of-fyb202-and-fyb201.
- Pearce IP. “Sandoz to ‘Minimise’ Phase 3 Biosimilar Pembrolizumab Trial Due to Regulatory Streamlining.” Pearce IP, 6 May 2025, www.pearceip.law/2025/04/30/sandoz-to-minimise-phase-3-biosimilar-pembrolizumab-trial-due-to-regulatory-streamlining.
- “FDA grants first ever waiver of clinical efficacy studies for monoclonal antibody biosimilars.” MedPath, 1 Sept. 2025, https://trial.medpath.com/news/5c50f3ccf4447df4/fda-grants-first-ever-waiver-of-clinical-efficacy-studies-for-monoclonal-antibody-biosimilars.
- Jorgensen, Paul. “Biosimilar Drugs Will No Longer Need Phase 3 Clinical Trials: Proposed Changes From Health Canada | Pharma in Brief.” Pharma in Brief, 20 June 2025, www.pharmainbrief.com/2025/06/biosimilar-drugs-will-no-longer-need-phase-3-clinical-trials-proposed-changes-from-health-canada.
- Heise, Tim, and J. Hans DeVries. “Biosimilar insulins: Narrative review of the regulatory framework and registration studies.” Diabetes, Obesity and Metabolism 27 (2025): 16-23.
- U.S. Biosimilars Surge: Clinical Trials, Regulatory Shifts, and the Path to Dominance. www.clinicalleader.com/doc/u-s-biosimilars-surge-clinical-trials-regulatory-shifts-and-the-path-to-dominance-0001.
- “Dr Reddy’s launches bevacizumab biosimilar Versavo in the UK”. GabiOnline.net, https://www.gabionline.net/biosimilars/news/dr-reddy-s-launches-bevacizumab-biosimilar-versavo-in-the-uk.
- Ituxredi EPAR I European Medicines Agency (EMA). Link: https://www.ema.europa.eu/en/medicines/human/EPAR/ituxredi.


