Preparing for Regulatory Audits at Mabion with Reliable Quality Systems
- Understanding the different regulatory audits approaches helps companies design quality systems that satisfy both EMA inspections and the more data-integrity-driven FDA audits.
- A robust pharmaceutical quality management system forms the foundation of regulatory readiness in biopharma companies.
- Continuous audit readiness enables companies to identify compliance weaknesses early and implement corrective actions before regulatory inspections occur.
Pharmaceutical Quality Management Systems: the basis for regulatory stability
The Pharmaceutical Quality System (PQS) described in the ICH Q10 provides a framework for managing quality throughout the entire lifecycle of a pharmaceutical product. It integrates regulatory Good Manufacturing Practice (GMP) requirements with biotech responsibility, ensuring that regulatory department actively supports quality objectives. Within this system, structured processes such as change control, supplier qualification, data integrity system, deviation handling, Corrective and Preventive Actions (CAPA), batch record review, and process performance monitoring enable organizations to identify and control risks that may affect product quality.1
The PQS also incorporates quality risk management principles from ICH Q9, which allow companies to evaluate potential risks and implement appropriate control strategies. By continuously monitoring process performance and product quality, the system enables organizations to improve operations, maintain regulatory compliance, and ensure consistent delivery of safe and effective medicines.2
Regulatory audit preparation should be a natural extension of everyday quality management. When operations actively reviews quality metrics, supports continuous improvement, and ensures proper training of personnel, the organization develops a culture of inspection readiness. This culture ensures that operational processes, documentation, and knowledge remain aligned with regulatory expectations at all times, which significantly increases the likelihood of a successful audit outcome.
Strategic Regulatory Audits Preparation
Strategic preparation for regulatory inspections begins long before auditors arrive on site. The process typically involves systematic internal audits, risk assessments, and gap analyses. These activities are designed to identify areas where procedures or practices may deviate from regulatory standards. When these evaluations are conducted thoroughly, they reveal weaknesses. These weaknesses may relate to documentation control, data integrity, validation practices, or operational consistency. Identifying such weaknesses early allows companies to implement corrective and preventive actions. These actions strengthen compliance and reduce the risk of regulatory observations.
Documentation Review and Filling Gaps in Procedures
A documentation review should begin with the establishment of a structured audit readiness program within the organization. Such a program ensures that regulatory preparedness is maintained continuously rather than addressed only when an inspection is announced. Companies typically implement regular internal audits, simulated regulatory inspections, and systematic evaluations of their quality system against current GMP requirements. These QA activities create a proactive environment in which documentation and procedures are regularly challenged, verified, and improved.
Routine internal audits form the backbone of this readiness program because they allow organizations to examine whether documentation truly reflects operational practice. During these audits, quality specialists review key quality documents and operational records to confirm that processes are being executed according to approved procedures.
Simulated regulatory inspections, often called mock inspections, replicate the structure and expectations of EMA or FDA audits, allowing companies to evaluate how easily documentation can be retrieved and how clearly it demonstrates compliance. These exercises help identify weaknesses in documentation systems before they become regulatory findings.3
An essential component of documentation review is a formal gap analysis against applicable GMP regulations and industry guidelines. This analysis examines whether company procedures and records fully meet regulatory expectations and whether any missing or outdated documents could create compliance risks. The gap analysis typically includes a systematic review of standard operating procedures, detailed inspections of batch manufacturing documentation, verification of validation records, and assessment of how data integrity principles are applied within documentation systems.
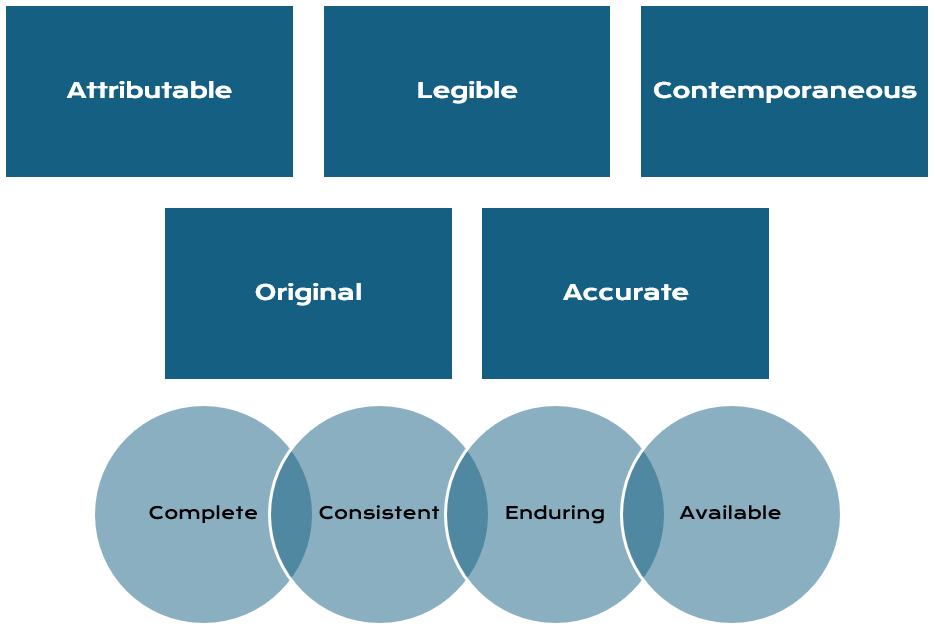
During this process, companies must ensure that all documentation meets strict regulatory standards for completeness and traceability. Documents must be properly approved, clearly version-controlled, and easily retrievable to demonstrate that procedures are followed consistently. At the same time, documentation systems must comply with data integrity principles such as ALCOA+, ensuring that information is attributable, legible, contemporaneous, original, accurate, complete, consistent, enduring, and available.4
Particular attention should be given to quality documentation, which forms the structural backbone of the pharmaceutical quality system. Core documents such as the Quality Manual, Site Master File (SMF), and Validation Master Plan (VMP) must accurately describe the organization’s quality framework, facility operations, and validation strategies.
These documents provide regulators with a high-level understanding of how the company manages compliance and quality risks. These documents must be current, internally consistent, and aligned with operational practices. This is essential for demonstrating the maturity of the quality management system. Organizations must maintain detailed operational documentation. This documentation should reflect the execution of manufacturing and quality processes. Standard operating procedures must clearly describe how activities are performed. Batch manufacturing records must provide a complete and traceable account of each production batch. During documentation reviews, companies verify that these records accurately reflect approved procedures. They also check that the records contain all required signatures, timestamps, and supporting data.
Personnel Training and Interview Preparation during Internal Audits and Mock Inspections
Staff training and preparation interviews during internal audits and mock inspections are designed to ensure that employees clearly understand both their operational responsibilities and the regulatory expectations associated with them. Training typically begins with reinforcing knowledge of GMP principles, relevant standard operating procedures (SOPs), and the structure of the company’s quality management system. Employees learn how their daily activities contribute to product quality and regulatory compliance, which helps them explain their work confidently and accurately during auditor interviews.
During internal audits and simulated inspections, personnel are often asked questions similar to those posed by regulatory inspectors. These exercises allow employees to practice explaining procedures, demonstrating how documentation is maintained, and showing where relevant records are stored.5
As training progresses, employees learn effective communication practices for interacting with auditors. They are encouraged to provide clear, factual answers, refer to written procedures when necessary, and avoid speculation or providing information outside the scope of the question. This preparation ensures that during a real regulatory inspection, employees can demonstrate that the organization’s quality procedures are consistently applied.
Proper Conduct during Regulatory Audits and Post-Audit Activities
Proper conduct during the inspection is essential. When inspectors arrive on site, the audit typically begins with an introductory meeting in which the company presents its structure, operational scope, and quality management framework. A clear and professional presentation establishes the context for the inspection and demonstrates that the organization approaches regulatory oversight with openness and preparation.6
As the inspection progresses through facility tours, document reviews, and interviews with employees, the organization must maintain disciplined communication and document management practices. Inspectors typically review drug manufacturing zone, QC laboratories, and data systems. Personnel should respond to questions accurately and refer to approved documentation whenever possible. Employees should avoid speculation, over-explaining, or providing unnecessary information.7
Following the completion of the inspection, regulatory auditors typically summarize their observations during a closing meeting. Possible audit outcomes:
- No observations
- Minor observations
- Major observations
- Critical findings
Organizations should listen carefully, ask clarifying questions, avoid defensive responses, and begin CAPA planning immediately after the audit.8
Differences Between EMA and FDA Regulatory Audits
Innovative biologics are subject to particularly rigorous regulatory review. Their complex manufacturing processes and biological nature make product quality highly sensitive to even minor deviations. Small levels of microbial, viral, or cross-contamination may have serious consequences for patients. These consequences may include severe adverse reactions or even death, both during clinical trials and after market approval. For this reason, EMA and FDA inspectors pay close attention to contamination control strategies, aseptic processing, raw material traceability, and environmental monitoring. They also assess the robustness of quality systems used to protect patient safety.
Both the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) enforce strict pharmaceutical manufacturing standards, their inspection approaches reflect differences in regulatory frameworks and operational focus. EMA inspections are typically conducted by national competent authorities within the European Union and are based on the EU GMP guidelines. FDA inspections, by contrast, are grounded in the current GMP regulations outlined in Title 21 of the Code of Federal Regulations.9
FDA regulatory audit frequently focus on the traceability of raw data and the integrity of electronic records, with inspectors carefully examining audit trails and system controls to confirm that data have not been manipulated. They often ask direct technical questions and may interview operators extensively. Compared to EMAs, inspectors are more investigative, focusing on weaknesses in the system.10
EMA regulatory audit often emphasize the effectiveness of the pharmaceutical quality system and the role of the Qualified Person in ensuring batch certification and product release. EMA inspectors ask more structured inspection plan with strong emphasis on QMS effectiveness. Focus areas include QP responsibilities, quality oversight and manufacturing authorization.7
Because of these differences, biotechnology companies preparing for international regulatory oversight must ensure that their quality systems satisfy both regulatory perspectives. This approach helps organizations meet the expectations of both the EMA and the FDA. Organizations can achieve this by aligning their internal practices with the expectations of both authorities. Doing so strengthens global regulatory compliance and reduces the risk of inspection findings.
FAQ
Prepared by:

Marketing Specialist
References
- European Medicines Agency. ICH Q10 Pharmaceutical quality system. 2008.
- European Medicines Agency. ICH Q9 Quality risk management. 2023.
- Ojo-Imoukhuede L. Implementing a QA-Driven Audit Simulation Program to Improve Regulatory Readiness in Biopharmaceutical Manufacturing: A Mixed-Methods Study. J. Pharm. Sci. 2025.
- Sembiring MH, Novagusda FN. Enhancing Data Security Resilience in AI-Driven Digital Transformation: Exploring Industry Challenges and Solutions Through ALCOA+ Principles. Acta Inform Med. 2023; 32(1): 65-70.
- Grzesiak L, Ulrych W. How auditor-oriented employee performance management affects internal audit effectiveness by reducing its constraints. Humanities and Social Sciences. 2024; 31(4): 99-121.
- Al Azawei A, Loughrey K, Surim K, Connolly ME, Naughton BD. The management of good manufacturing practice (GMP) inspections: a scoping review of the evidence. Front Med (Lausanne). 2025; 12: 1687864.
- European Medicines Agency. Compilation of Union procedures on inspections and exchange of information. 2024.
- Prasanna Gayathri B, Kamaraj R. Pharmaceutical Inspection Co-operation Scheme: An Overview. Cureus. 2024; 16(9): e69043.
- Tuszyner A, Małecka A. Similar but not the same: an in-depth look at the differences between EMA and FDA. Mabion Science Hub. 2024.
- Tran R, Fraser G, Fisher AC, Lee SL, Boam A, Tsinontides S, Maguire J, Yu LX, Rosencrance S, Kozlowski S, Henry D. A network of regulatory innovations to improve FDA quality assessments of human drug applications. Int J Pharm X. 2024; 7: 100239.


