Mass spectrometry in peptide and protein analysis
Analytics, Drug substance, Proteins
- The development of key mass spectrometry (MS) innovations in the 1980s, coincided with the emergence of recombinant therapeutic proteins. Today, MS is a cornerstone of biologics characterization, with nearly all applications incorporating MS data to assess drug’s quality.
- Tailoring the analytical strategy, including ionization technique, fragmentation method and mass analyzer type, to the analyte’s properties is crucial to ensure sensitivity, accuracy and compliance with regulatory standards.
- Integration of MS with other analytical devices enhances sensitivity and resolution of this method required for more complex biologics. As MS workflows become more sophisticated, advanced software tools and compliant data acquisition systems are essential for managing complex datasets and ensuring robust, reproducible results.
- The optimization of MS is a laborious task, however if properly handled, the MS method can provide data of unparalleled quality and precision.
Introduction
The 1980s marked a significant breakthrough for medical biotechnology and mass spectrometry. The approval of recombinant insulin in 1982, the first therapeutic protein for human use derived from recombinant DNA, coincided with significant advancements in mass spectrometry. Around that time, newly developed ionization techniques, specifically electrospray ionization (ESI) and matrix-assissted laser desorption/ionization (MALDI), were first applied to the analysis of biomolecules. In 1988, two independent teams, led by Tanaka1, and Hillenkamp & Karas2, demonstrated the potential of MALDI for analyzing large proteins. Shortly after, in 1989, Fenn and co-workers published a seminal paper in Science illustrating the ability of electrospray to ionize complex biomolecules.3 These breakthroughs in ionization techniques ushered in a new era of protein structural analysis, enabling the comprehensive characterization of therapeutic proteins.
The studies conducted by the U.S. Food and Drug Administration (FDA) have underscored the importance of mass spectrometry in the characterization of therapeutic proteins. An analysis of Biologics License Applications (BLAs) for 80 therapeutic proteins approved between 2000 and 2015 revealed that only one submission did not include MS data.4 A follow-up study covering 93 BLAs approved between 2016 and 2020 found that all included MS analysis.5 The eight most commonly assessed quality attributes using MS were:
- Amino acid sequence,
- Molecular mass,
- Oxidation,
- Disulfide bonds,
- Deamidation,
- Glycosylation,
- N- and C-terminal sequence variants,
While the attributes were consistent across both studies, their relative ranking varied in terms of prevalence. The increasing adoption of MS-based methods for therapeutic protein characterization was particularly evident in the 2000-2015 dataset. During this period, MS was used to confirm amino acid sequences in over 91% of applications. The use of MS for molecular mass determination rose from 83% to 97%, while disulfide bond mapping increased from roughly 50% to over 90% by 2011. Glycosylation analysis via MS ranged between 62% to 76%, and characterization of N- and C-terminal variants, mainly focusing on pyroglutamate formation and lysine clipping, increased from approximately 40% to 70%, Additionally, the use of MS to detect deamidation and oxidation rose from 17–25% in the early 2000s to 76–80% by the end of the analyzed period. Beyond individual attributes, the number of MS-assessed attributes per BLA grew substantially: from an average of two in 2000 to 11 by 2015. Notably, one BLA from 2015 included MS analyses for 18 distinct attributes. On average, each BLA contained MS data for just over eight attributes; this average exceeded 9.5 for antibodies and was slightly above six for general proteins. This trend reflects the growing availability and accessibility of high-resolution mass spectrometers, enabling deeper characterization.4,5
The first sections of this paper discuss the fundamental principles of mass spectrometry that underpin the analysis of peptides and proteins. Although MS can accommodate a broad range of analytes differing in polarity, molecular weight, and stability, each class of molecules necessitates specifically tailored instrumentation. Therefore, selecting appropriate equipment and optimizing analytical parameters are critical steps for reliable data acquisition. Following this, the major applications of mass spectrometry in therapeutic protein characterization are explored, grouped into three core methodological approaches: bottom-up, middle-down, and top-down. The top-down strategy analyzes intact proteins, the middle-down approach targets large subunits (25–100 kDa), and bottom-up analysis involves peptide fragments generated by enzymatic digestion. The subsequent section addresses key challenges encountered in mass spectrometry-based workflows, spanning sample preparation, instrument performance, and data interpretation. Finally, the paperpresents advanced and integrated analytical approaches. such as ion mobility–mass spectrometry (IM-MS), two-dimensional liquid chromatography–mass spectrometry (2D LC-MS), nanoLC-MS, and capillary electrophoresis–mass spectrometry (CE-MS), that offer enhanced resolution, sensitivity, and structural insights for complex biologics.
Mass spectrometry techniques in protein and peptide analysis
The selection of an appropriate mass spectrometer is a critical determinant of analytical success and must be tailored to the physicochemical properties of the analyte, such as its polarity, molecular weight, volatility and stability. These factors dictate the need for instrument components to be specifically matched to the analyte’s characteristics. Key elements of mass spectrometry systems, along with a comparison between single-analyzer instruments and tandem mass spectrometry (MS/MS) systems, are summarized in Figure 1. For the analysis of large biomolecules, including therapeutic proteins, dedicated ionization methods are required to ensure efficient ion formation. Furthermore, the application of suitable fragmentation techniques is essential for accurate amino acid sequence analysis, which is fundamental to understanding protein structure and biological function.
The selection of an appropriate mass analyzer is equally crucial, as it significantly impacts resolution and accuracy – parameters that collectively influence the reliability of the results. In addition, the mode of data acquisition and the capabilities of the data processing software must be carefully evaluated, as these directly impact the interpretation and usability of the results. Consequently, equipment selection must be made with deliberate attention to the specific analytical objectives and intended applications.
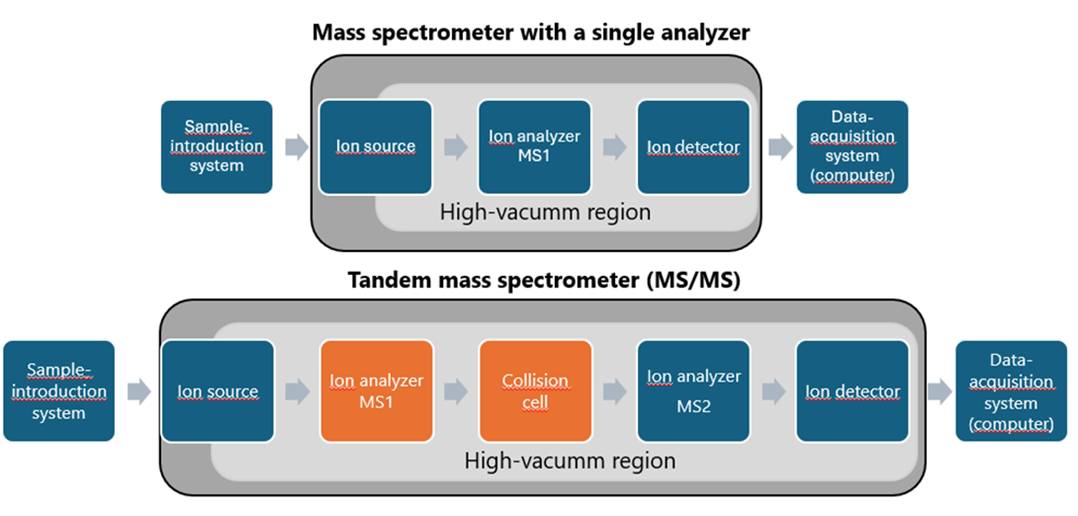
A wide range of high-performance mass spectrometers is available on the market, each offering distinct capabilities depending on the manufacturer and system configuration The choice of system should align closely with analytical goals and regulatory requirements. For example, native protein analysis typically requires instruments with a broad m/z detection range, a specification that is less critical for denatured proteins or peptide-based analyses. For biopharmaceutical laboratories, compliance with regulatory frameworks, such as 21 CFR Part 11, is often mandatory, whereas such considerations may be less relevant in academic or exploratory research settings.
Aligning instrument capabilities with the intended analytical purpose also supports cost-effectiveness by preventing unnecessary expenditure on features that are not required. This section provides an overview of key techniques in protein and peptide mass spectrometry, including sample introduction, ionization, and fragmentation strategies, as well as a comparison of commonly used mass analyzers. Additionally, available data acquisition modes and advanced software tools for data analysis are discussed to guide optimal system selection and method development.
Sample introduction
For the analysis of proteins and peptides, which are polar, non-volatile molecules soluble in aqueous solutions, liquid chromatography is the preferred method for introducing samples into a mass spectrometer equipped with an ESI source. This approach allows for the effective separation of peptides and proteins, facilitating both qualitative and quantitative mass spectrometry analysis. High-performance liquid chromatography (HPLC) and ultra-high-performance liquid chromatography (UHPLC) are widely used tools in this context and are applicable to both complex protein mixtures and individual analytes. Depending on the specific analytical objectives, laboratories may also employ nano-flow liquid chromatography (nanoLC) or capillary electrophoresis (CE). These techniques are particularly valuable for the analysis of samples containing low concentrations of multiple proteins. Their low flow rates improve ionization efficiency and sensitivity, resulting in lower detection limits. The integration of mass spectrometry with CE, nanoLC, and two-dimensional liquid chromatography (2D-LC) represents a powerful strategy for enhancing analytical performance. These advanced approaches will be discussed in greater detail in the section Advanced analytical strategies for peptide and protein characterization.
Ionization
Multiple ionization techniques are available for peptides and proteins, with selection dependent on analyte characteristics and the analytical goal. Techniques such as field desorption (FD) and fast atom bombardment (FAB) can be used for ionization of peptides and small proteins.6–8 However, the analysis of large, non-volatile biomolecules became feasible only with the advent of electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI)—now the two most commonly used techniques for protein and peptide ionization.
In MALDI, protonated molecules are generated by coprecipitating an excess of matrix solution with the analyte. A small volume of this mixture is pipetted onto a metal target plate and dried at room temperature. Nanosecond laser pulses, typically from UV nitrogen lasers (337 nm or occasionally 335 nm) and sometimes IR lasers, are then used to irradiate the resulting “solid solution”. The matrix absorbs the laser energy, leading to vibrational excitation and disintegration of the “solid solution”, forming clusters of analyte molecules surrounded by neutral matrix species. As the matrix evaporates, excited analyte molecules remain ionized and are electrostatically guided into the mass analyzer. Ion formation can occur through protonation (M + H)⁺ or deprotonation (M – H)⁻, depending on the mode. Additionally, ionized analyte can form via the adduction with small ions, such as Na⁺, K⁺, acetate, or ammonium, present in the MALDI solution.9 MALDI-TOF MS offers several advantages, including high buffer tolerance, clean spectra dominated by singly charged ions, and rapid, high-throughput analysis with minimal sample and reagent consumption. However, its application to intact glycoproteins is limited by poor ionization efficiency, fragmentation of labile groups (e.g., sialic acids, GlcNAc), and the matrix-dependent variability in spectral quality.10
In contrast to MALDI, electrospray ionization (ESI) operates at atmospheric or near-atmospheric pressure. In ESI, a dilute analyte solution in a polar, volatile solvent is infused through a stainless-steel capillary at low flow rates (typically 1–20 µL/min). A high voltage (2–6 kV) is applied to the capillary tip, creating a strong electric field that disperses the solution into a fine aerosol of highly charged droplets. A coaxial sheath gas (dry nitrogen) enhances nebulization and directs the spray toward the mass spectrometer. As the charged droplets evaporate, they decrease in size, releasing charged analytes that pass through a sampling cone or the heated capillary orifice into the mass spectrometer analyzer, which operates under high vacuum.11 ESI methodology can be performed either in positive- or negative-ion mode, depending on the proton affinity of the analyte. Protein and peptide analyses are typically conducted in positive-ion mode, which generally provides higher ion yields, although negative-ion analysis is also feasible. These analytes frequently carry multiple charge states, with their distribution influenced by the number and position of basic amino acid residues. The overall ion yield is further influenced by the hydrophobicity of the peptide/protein and efficiency of charge acquisition.12
Whereas hard ionization techniques such as electron ionization (EI) induce extensive in-source fragmentation, useful for small-molecule structure evaluation, soft ionization methods like ESI and MALDI minimize in-source fragmentation. This makes tandem mass spectrometry (MS/MS) essential for detailed structural analysis. MS/MS introduces a second stage of fragmentation, allowing targeted dissociation of selected precursor ions. This selective and controlled fragmentation enhances analytical specificity and provides defined fragment ion patterns, which are crucial for accurate peptide sequencing.
Fragmentation
Collision-induced dissociation (CID) is the most commonly used ion-fragmentation technique in mass spectrometry. It is also the predominant method for analysing peptides and proteins. In CID, accelerated precursor ions collide with neutral gas molecules, typically nitrogen or argon, converting a part of ions’ translational energy into internal energy and causing their fragmentation.
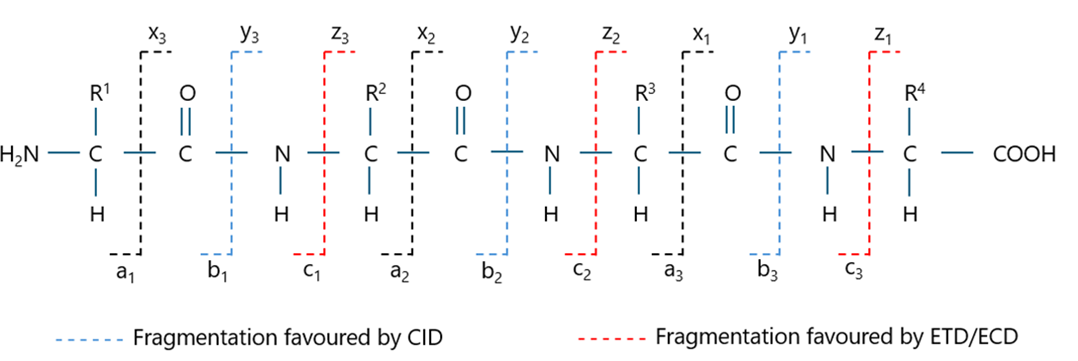
CID primarily breaks the amide bonds (CO-NH) in peptides, generating b- and y-ion fragments as presented in Figure 2.13 In the late 1990s and early 2000s, two alternative fragmentation methods were developed: electron-capture dissociation (ECD) and electron-transfer dissociation (ETD), respectively. In ECD, a multiply protonated peptide or protein cation captures a low-energy electron, causing fragmentation at the N-Cα bond and producing c- and z-ion series. In ETD, electrones from radical anions are transferred to the multiply protonated peptides, also producing c- and z-ion fragments.14 ECD generates more abundant MS/MS spectra and provides improved sequence coverage compared to CID, however its use is generally limited to expensive FTICR instruments. On the other hand, ETD can be performed on more affordable mass spectrometers. Compared to CID, ETD is particularly useful for analyzing large peptides, proteins, and labile post-translational modifications (PTMs). The complementarity of CID and ETD techniques is also emphasized.15
Mass analyzers
All mass spectrometers operate by measuring the mass-to-charge ratio (m/z) of ions, however their precision depends heavily on the type and characteristics of the instrument. Two critical parameters used to evaluate mass spectrometers are mass accuracy and resolving power.
Mass accuracy is defined as the difference between the observed and true mass, normalized to the true mass, and is typically expressed in parts per million (ppm). It is calculated using the formula:
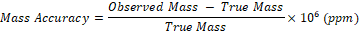
Resolving power (R), on the other hand, is defined as the ratio of the m/z value of a peak to its full width at half maximum (FWHM):
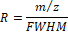
Higher resolving power results in narrower and better-defined peaks, significantly improving the instrument’s ability to differentiate ions with very similar m/z values. This is particularly important for resolving isotopic envelopes and distinguishing between compounds with closely spaced mass differences, where spectral overlap can lead to misinterpretation or quantification errors.
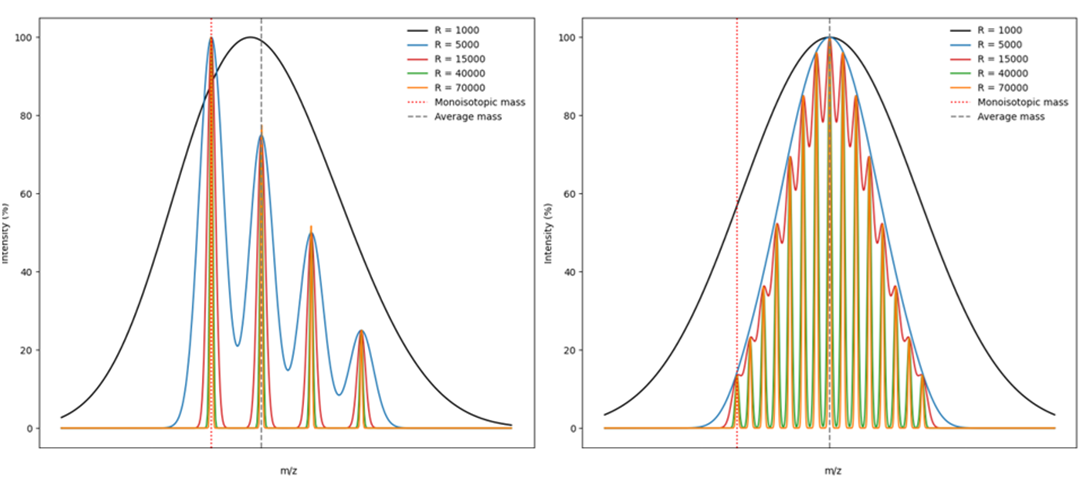
Figure 3 presents hypothetical mass spectra, illustrating the impact of resolution on spectral quality. The comparison clearly shows that low-resolution instruments produce merged isotopic peaks, whereas high-resolution instruments allow for clean separation of isotopic signals, facilitating accurate identification and characterization.
The most commonly used high-resolution mass spectrometers for peptide and protein analysis are equipped with either time-of-flight (ToF) or Orbitrap mass analyzers. Notably, tandem mass spectrometry (MS/MS) systems are considered a standard in proteomic and biopharmaceutical applications. These devices typically combine two mass analyzers: one isolating precursor ions for fragmentation and the other performing mass analysis of the isolated ions. The former is most often a quadrupole, while the latter is a high-resolution analyzer such as ToF or Orbitrap.
The quadrupole mass analyzer consists of four parallel metal rods with hyperbolic or circular cross-sections, symmetrically arranged around the central axis. Ions generated in the ion source are directed into this axis. Each opposing pair of rods is connected to a combination of radiofrequency (RF) and direct current (DC) voltages, with adjacent rods operating at opposite RF phases. This configuration generates an oscillating electric field within the quadrupole.
When ions enter the quadrupole, their trajectory is influenced by the applied RF and DC fields. For a given set of voltage parameters, only ions within a narrow m/z window exhibit stable trajectories and are transmitted through the analyzer. Ions outside this stability region become destabilized, strike the rods, and are neutralized. By adjusting the RF and DC voltages, the quadrupole can be tuned to selectively transmit ions of specific m/z values, effectively functioning as a mass filter. This ion-filtering property makes quadrupoles essential in tandem MS workflows, where they serve as the first stage in precursor ion selection prior to fragmentation and high-resolution analysis.16
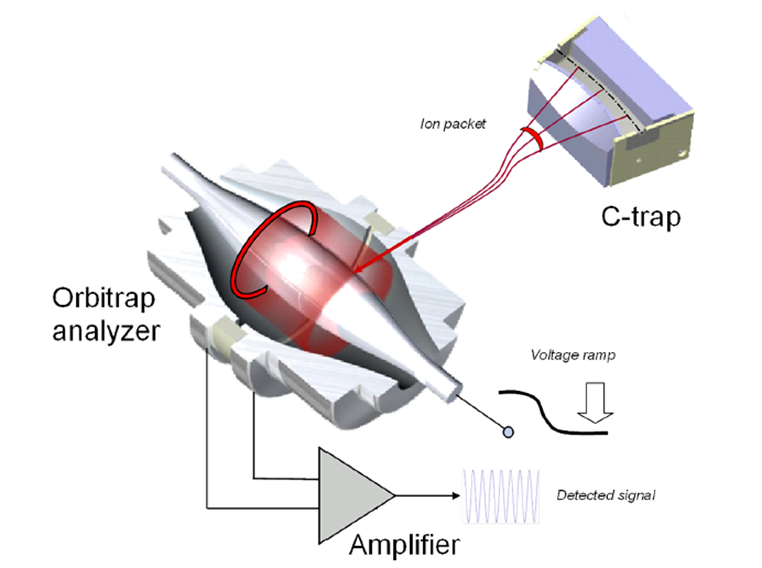
A mass spectrometer with an Orbitrap analyzer combines high-resolution power with excellent sensitivity. The analyzer consists of three electrodes: two cup-shaped outer electrodes that are electrically isolated and face each other, and a central spindle-shaped electrode. When a voltage is applied between the central and outer electrodes, an electric field is generated. This field is linear along the axis and results in purely harmonic oscillations in this direction. At the same time, ions are strongly attracted to the central electrode by the radial component of the field. Ions are injected into the space between the central and outer electrodes. The radial electric field bends their trajectory toward the central electrode, while their tangential velocity creates an opposing centrifugal force. With the right parameters, ions follow a nearly circular spiral within the trap. Simultaneously, the axial electric field, caused by the conical shape of the electrodes, induces harmonic oscillations along the axis. The outer electrodes act as receiver plates, detecting the image current generated by these oscillations. This digitized image current is then Fourier-transformed from the time domain to the frequency domain and ultimately converted into a mass spectrum.17 A schematic of Orbitrap mass analyzer is provided in Figure 4.
The operartion of ToF analyzer is based on measuring the travel time of ions through a flight tube. Ions are accelerated by applying a high voltage and travel a specific distance through analyzer to the detector. Assuming that all ions entering the analyzer possess the same initial kinetic energy, their velocities vary according to their masses – lighter ions move faster, while heavier lag behind. Since all ions travel across the same distance within the spectrometer, their time of flight is proportional to their mass. Consequently, heavier ions reach the detector later than lighter ones and the measured time of flight can be used to calculate the mass-to-charge ratio (m/z).
Acquisition modes
The choice of MS acqusition mode is determined by the purpose of the performed analysis. There are three main MS data acquisition modes:
- Full-scan acquisition,
- Data-dependent acquisition (DDA),
- Data-independent acquisition (DIA).
Both DDA and DIA can be divided into targeted and untargeted modes.
Full-scan mode is a scanning method in which the mass analyzer scans all ions within a specified mass range (m/z) without selecting specific ions for fragmentation. It provides general information about the sample, recorded mass-to-charge ratios (m/z) and their relative abundances.
In DDA, the experimental scheme typically involves the selection, accumulation, and fragmentation of precursor ions based on the data from MS1 survey scan. The targeted DDA variant, in which user can select for fragmentation and analysis specific precursor ions (predefines m/z values) is useful when looking for specific compounds. In untargeted version there is no ion pre-selection and the device automatically selects precursors for MS/MS based on signal intensity (the strongest signals within a given time window). Importantly, untargeted DDA does not require prior knowledge of sample’s composition. It prioritizes and provides detailed information on the most abundant compounds in the sample, ignoring contaminants that may be present in low amounts.
In DIA, instead of selecting ions based on their intensity, all ions within the defined m/z windows are fragmented, and MS/MS data is collected simultaneously for all of them. Like DDA, DIA can also be either targeted or untargeted. The two targeted DIA methods are SRM/MRM (Selective Reaction Monitoring/Multiple Reaction Monitoring) and PRM (Parallel Reaction Monitoring). In the MRM strategy, the first analyzer selects precursor m/z corresponding to the peptide(s) of interest for fragmentation. The resulting fragment ions are then monitored with the use of second analyzer. The specific combinations of m/z values associated with the peptide precursor and fragment ions are known as transitions. This method is commonly performed using a triple-quadrupole mass spectrometer (QQQ), which is regarded a an effective tool for quantitatve analysis due to its high sensitivity and beneficial noise-to-signal ratio. In contrast to MRM, PRM (Parallel Reaction Monitoring) combines the use of a quadrupole and a high-resolution mass analyzer (such as Q-ToF or Q-Orbitrap). Rather than monitoring specific fragments in QQQ, PRM captures all fragment ions from the selected precursor with high mass accuracy. Like MRM, PRM offers excellent selectivity and quantitative reliability, while providing greater flexibility for post-acquisition data analysis.
For untargeted workflows, approaches like uDIA or SWATH (Sequential Window Acquisition of All Theoretical Mass Spectra) are employed. These methods fragment all ions within predefined m/z windows across the full mass range, without pre-selecting specific precursors. This versatile approach is particularly suited for discovery-based research and profiling of unknown analytes, offering broad coverage and high data completeness.18
Software
The contemporary bioanalytical software offers powerful tools for identifying and quantifying peptides and proteins, including their modifications. The availability of robust data analysis and visualization modules, capable of processing large and complex datasets at an unprecedented speed, gave an incredible boost to the biopharmaceutical research. In the case of mass spectrometry, the IT revolution resulted in the development of huge spectral libraries and protein databases, which allowed scientists from all around the world to quickly search the obtained spectra and identify peptides with exceptional accuracy. The currently available software also supports advanced analysis of the protein structure e.g., offering in-depth look into its glycosylation profile. Also, using advanced deconvolution algorithms it can process both isotopically resolved and unresolved mass spectra, delivering accurate molecular weight determination.
Importantly, an increasing number of IT companies introduce solutions that help maintain the compliance of data acquisition and processing with federal regulations, including FDA 21 CFR Part 11. Functionalities, like tracking all actions and enforced logins, are built-in in order to ensure data integrity, security and reliability.
Mass spectrometry for peptide analysis (bottom-up approach)
Amino acid sequence
For therapeutic proteins, the amino-acid sequence is precisely defined and must be verified during the development process. It is also essential to demonstrate the consistency of this sequence across production batches to ensure product’s quality. In the case of biosimilars, regulatory authorities require confirmation of sequence identity between biosimilar candidate and the reference drug with single-amino-acid resolution. Achieving this level of precision is critical to demonstrate molecular comparability and support regulatory approval.19,20
Technological advancements in mass spectrometry along with the widespread availability of tandem mass spectrometers, particularly high-resolution ones like Q-ToF and Q-Orbitrap, have established these platforms as the preferred tools for determining the amino acid sequences of proteins. Sequence confirmation is typically performed using the bottom-up approach, which involves enzymatic digestion of the protein into peptides, followed by chromatographic separation and MS/MS analysis. Among fragmentation techniques, CID (collision-induced dissociation) is the most widely employed. Under CID conditions, peptides predominantly fragment at the CO-NH bond as illustrated in Figure 2. The resulting b and y ions provide the basis for peptide identification and accurate sequence verification. To ensure robust sequence confirmation, stringent evaluation criteria must be applied. Two critical parameters are: (1) high mass accuracy, demonstrated by a low relative mass error (typically expressed in parts per million, ppm), and (2) comprehensive sequence coverage, ensuring that fragment ions confirm the identity of each amino acid residue in the peptide.
Despite the capabilities of modern MS systems, several challenges can arise during sequence analysis. These are addressed in the following section: Common Challenges in Mass Spectrometry-Based Analysis.
Post-translational modifications
Post-translational modifications (PTMs) of proteins are chemical changes to their amino acid residues that occur during gene expression, purification, and long-term storage, influenced by various chemical and enzymatic processes. The impact of a specific modification on the drug’s efficacy and safety largely depends on its type and location. For example, modifications of residues in the complementarity-determining region (CDR) of an antibody can negatively impact binding to target antigens, while those occurring in the constant domains of the Fc typically have minimal effects.21 Therefore, precise identification of the type and location of modifications within the protein is extremely important. Mass spectrometry enables the identification of post-translational modifications and the precise localization of modified amino acid residues within a protein sequence. The type of modification is indicated by the mass difference between the modified and unmodified residues, depending on the nature of the modification. Quantification is typically based on measuring the area under the peak in the Extracted Ion Chromatogram (EIC) for the relevant m/z values. Based on the area under the peaks for the same peptide fragment in its modified and unmodified forms, the percentage of modification at a specific residue within the sequence can be determined. The analysis of modifications involves various challenges, which will be discussed in the section Common Challenges in Mass Spectrometry-Based Analysis.
The ability to identify and quantify post-translational modifications using mass spectrometry (MS) can be utilized in comparability studies, allowing for effective comparison of different batches of drug products and in similarity studies to compare a biosimilar candidate with the reference. Additionally, it plays a crucial role in stability studies, by assessing how modification levels change over time under various storage conditions. Additionally, it can support supplementary research aimed at evaluating the susceptibility of specific amino acid residues to modifications and the impact of these modifications on biological function. For example, incubating a protein with an oxidizing agent, such as hydrogen peroxide, followed by analysis using MS, allows researchers to determine which amino acid residues are most prone to oxidation. Furthermore, introducing artificial modifications at defined levels, established based on MS data and followed by biological analyses, enables the investigation of how specific levels of modification affect various biological functions.
A post-translational modification that deserves particular attention is glycosylation. It is one of the most common post-translational modifications of proteins, playing a crucial role in their biological activity and influencing their pharmacokinetics and immunogenicity. The glycosylation pattern depends on various factors, including the host cell line and cell culture conditions.22 Due to its biological significance, glycosylation is extensively studied. Research focuses on the degree of glycosylation site occupancy and the glycosylation pattern, including the types and relative abundance of specific glycans in the protein, as well as the quantity of individual monosaccharides that constitute the glycans. In glycan analysis, techniques like LC/CE with fluorescent detection, following prior labeling with fluorescent tags, are commonly used. However, mass spectrometry also plays a significant role in glycosylation studies, providing an orthogonal or complementary approach to other methods.
Glycopeptide analysis offers a precise method for characterizing the glycosylation profile, as glycans remain bound to their respective amino acid residues. This is especially valuable for proteins with multiple glycosylation sites, enabling site-specific glycan identification. The technique yields both qualitative and quantitative insights, following a workflow similar to that used for other post-translational modifications. Furthermore, LC-MS/MS analysis of glycopeptides allows for the simultaneous identification of amino acid sequences and glycan structures, within the same run.
Additionally, for each glycosylation site, it is possible to determine its site occupancy level. This parameter is defined as the ratio of the glycosylated antibody to the sum of its glycosylated and non-glycosylated forms. Because precise semi-quantitative analysis cannot be conducted on the intact protein, a bottom-up approach is employed. In this method, peptide fragments containing the glycosylation site, both glycosylated and non-glycosylated are generated and analyzed using LC-MS. However, the presence of an attached glycans can significantly affect peptide ionization efficiency. To overcome this problem, the antibody is subjected to enzymatic deglycosylation. This process removes the glycan and results in the conversion of asparagine (Asn) to aspartic acid (Asp), which can be detected as a distinct mass shift. Since the two peptides differ by only one amino acid and these residues have very similar physicochemical properties, ionization efficiency is assumed to be unaffected.
The glycosylation profile can be also obtained through glycan-based analysis. This process involves the release of glycans from glycoproteins, which are then typically labeled with a fluorescent tag and analyzed using LC/CE with fluorescence detection. This method provides insights into the glycosylation profile and the percentage of individual glycoforms. Fluorescence detection ensures high precision in quantifying the percentage of glycoforms. If the glycoforms are identified and well-separated, their identification can be based on retention times. When there is uncertainty about the glycoforms present in a protein or if confirmation is needed, mass spectrometry (MS) can assist in their identification. This is feasible if the mobile phases and labeling reagents are chosen to ensure effective ionization of the labeled glycans. When these conditions are met, the same method can be applied using both fluorescence detection and MS. This allows for the identification of glycans at specific retention times on the column.
Mass spectrometry for protein analysis (top-down and middle down approach)
Molecular weight determination
Unlike bottom-up methods, the top-down approach analyzes the complete, unfragmented protein. Currently, this strategy is most commonly employed for determining the molecular weight of proteins. In contrast to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), high-resolution mass spectrometers offer significantly greater mass accuracy, with MW error usually below a few Da (0.01% error or lower), whereas SDS-PAGE typically yields molecular weight estimates with an error margin of 10%.23 This step provides critical insight into the sample’s composition. For instance, it allows for the assessment of protein heterogenity by detecting the presence of isoforms or post-translational modifications. In many cases, particularly in the analysis of therapeutic proteins where the theoretical protein mass is known, measured molecular weight can be directly compared to the expected value. Any discrepancies may be highly informative, potentially indicating N- or C-terminal truncations, or differences in post-translational modifications such as glycosylation. This comprehensive initial assessment is essential for verifying protein integrity, guiding quality control processes, and ultimately ensuring the safety and efficacy of biopharmaceutical products. An example of a protein mass spectrum before and after deconvolution is shown in Figure 5.
As the molecular weight of a protein increases, the number of incorporated heavy isotopes such as 13C and 15N also rises. Consequently, the isotopic distribution widens with increasing mass, forming an isotopic envelope, a series of mass spectral signals that, while differing in isotopic composition, share the same elemental formula. In the case of large proteins, the monoisotopic mass becomes harder to detect and thus, for large biomolecules like monoclonal antibodies, the average molecular mass is reported instead of the monoisotopic mass. However, in middle-down approach where protein subunits (for example, ~25 kDa light chain) are analyzed, high-resolution instruments can still resolve and report monoisotopic masses.
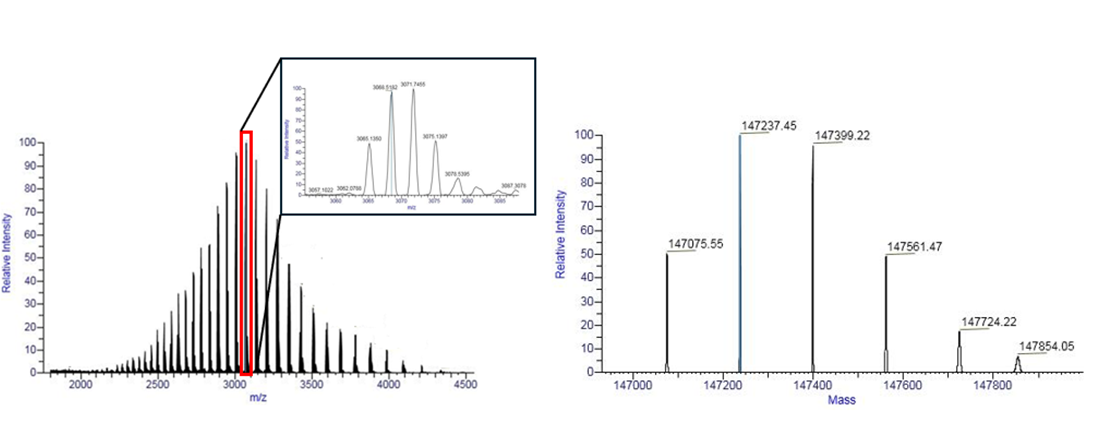
Before deconvolution, a broad charge envelope is observed due to multiple charge states of the protein. After deconvolution, distinct mass peaks corresponding to different glycoforms are visible, with mass shifts of 162 Da reflecting variations in glycan composition.
Measurement conditions exert a significant influence on the appearance of the mass spectrum. Denaturing conditions, which include organic solvents and non-neutral pH, disrupt non-covalent interactions and unfold protein structures, exposing additional protonation sites. This leads to a broader charge state distribution and higher average charge states in ESI-MS. Conversely, native conditions preserve the protein’s tertiary and quaternary structure, limiting accessible protonation sites to surface residues. As a result, native ESI yields ions with lower charge states and a narrower charge distribution. Importantly, the reduced charge states under native conditions shift the m/z values of protein ions toward higher values compared to their denatured forms.24
Ionization technique also plays a critical role in the appearance of the mass spectrum and the approach to molecular weight determination. Electrospray ionization (ESI) generates multiply charged ions, owing to the multiple protonation sites such as backbone amides and side-chain functional groups. As a result, ESI-MS spectra require deconvolution using dedicated software to convert m/z values into neutral mass values. In contrast, matrix-assisted laser desorption/ionization (MALDI) predominantly produces singly charged ions. Although low-intensity doubly or triply charged ions may occasionally be observed, the predominance of singly charged species greatly simplifies spectral interpretation and typically eliminates the need for deconvolution.25
Sequencing
In addition to being used for determining the molecular weight of entire proteins or their subunits, top-down and middle-down approaches are increasingly applied in protein sequencing, where intact proteins are directly fragmented within the mass spectrometer. In top-down MS workflows, fragmentation of intact protein is achieved using different fragmentation techniques such as: ECD, ETD, CID and HCD (Higher-energy Collisional Dissociation). These fragmentation methods are crucial for generating informative product ions, which can be then analyzed using high-resolution mass analyzers. Often, a combination of different fragmentation techniques is employed to maximize sequence coverage and enhance the quality of structural information. This strategy was first introduced in 1999 by Neil Kelleher, Fred McLafferty, and colleagues, who employed Fourier transform mass spectrometry (FT-MS) to sequence carbonic anhydrase B, a 29 kDa protein.26 Since then, interest in top-down sequencing has steadily grown, largely driven by significant technological advancements. Ongoing improvements in instrumentation and data analysis techniques continue to enhance the method’s importance in proteomics and therapeutic protein applications.
The top-down approach can be particularly valuable in proteomics, where the conventional bottom-up strategy complicates protein identification, since identical peptide fragments may originate from different isoforms or closely related proteins within the same family. In the context of targeted protein analysis, particularly in biopharmaceutical applications where confirming the exact protein sequence is essential, top-down approach offers a key advantage by eliminating the need for time-consuming, multi-step sample preparation. Despite its clear advantages, the top-down approach still faces several technical challenges. It requires high-resolution, highly sensitive instruments, such as Orbitrap or ToF mass spectrometers. Additionally, ion charge state distribution is broader for intact proteins, and electrospray ionization is generally less efficient compared to peptides, which can further reduce overall sensitivity. While continuous improvements are being made, achieving 100% sequence coverage remains difficult, particularly for large proteins (>50 kDa). Smaller proteins tend to yield better coverage, though even here, complete sequence characterization is rarely attained. One illustrative example is the light chain of adalimumab, a monoclonal antibody with a molecular weight of approximately 25 kDa. In a targeted nano-LC FT-ICR MS/MS experiment using both CID and ETD, researchers achieved 72% sequence coverage, demonstrating the method’s promising capabilities.27 However, continued technological progress in the field of mass spectrometry may soon enable complete sequence coverage of therapeutic proteins using the top-down approach.
Common challenges in mass spectrometry analysis
Although mass spectrometry technology has advanced significantly, analyzing biotherapeutic proteins using MS remains challenging throughout all stages, from sample preparation to the interpretation of results. Optimizing sample preparation protocols, selecting the most appropriate spectrometers, equipped with ionization and fragmentation techniques that match the analytical purpose, and choosing appropriate data acquisition and interpretation methods can encounter multiple obstacles that must be addressed. Furthermore, complying with rigorous regulatory requirements adds another dimension of complexity. This section delves into these challenges more thoroughly.
Sample preparation and matrix effect
LC–MS analyses are frequently challenged by matrix effects arising from co-eluting substances. These effects significantly impact key analytical parameters, including the detection limit, precision, and accuracy. The matrix effect, typically manifested as ion suppression, is commonly observed in analyses involving complex biological matrices. Studies have reported issues with reproducibility and accuracy when analyzing trace amounts of analytes in biological fluids, which hinder the detection of low-abundance compounds. Biopharmaceutical products are no exception; formulation components, such as polysorbate 80 (Tween 80®), are well-documented contributors to ion suppression.28 To overcome these challenges and improve method sensitivity and reliability, robust matrix clean-up procedures that effectively reduce background interference must be implemented.
Sample preparation can be an Achilles’ heel of the post-translational modifications’ analysis, as various artifacts may be introduced during this process. Careful attention must be paid prior to the analysis to ensure that the adopted protocol doesn’t modify the analysed molecules. Suboptimal or prolonged enzymatic digestion conditions (for example, elevated pH levels or lengthy incubation times) may result in deamidation, oxidation, and other structural changes, which can be misinterpreted as modifications occurring during protein production and storage.29
Structural impacts on ionization and fragmentation
Another important issue in quantitative MS-based peptide analysis is the variability in ionization efficiency between modified and unmodified peptides. While some post-translational modifications (PTMs) have minimal impact on ionization, others, such as glycosylation, can significantly reduce its efficiency. Evaluating the impact of specific modification on peptide ionization is crucial for accurate quantification. This can be achieved by analyzing equal concentrations of synthetic peptides with and without the modification. To accurately measure site occupancy, antibodies are typically deglycosylated enzymatically, which in addition to the full removal of glycans results in convertion of asparagine residues to aspartic acid. This process causes a measurable mass increase without altering ionization (the physicochemical properties of both residues are very similar), allowing for reliable quantification of deglycosylated and unglycosylated peptides.
One of the major pitfalls in amino acid sequence analysis is the ability to distinguish bertween leucine (Leu) and isoleucine (Ile), two isomers that share identical molecular mass and elemental composition. One effective strategy makes use of the distinct cleavage preferences of chymotrypsin, which favors Leu and aromatic residues over Ile, cleaving the latter with up to 40 times lower efficiency. Since leucine and isoleucine cannot be reliably differentiated in MS/MS spectra obtained via collision-induced dissociation (CID), alternative fragmentation methods must be employed. A common approach involves combining multiple fragmentation techniques, such as electron transfer dissociation (ETD) and higher-energy collisional dissociation (HCD). The ETD is first used to generate z-ions from doubly or triply charged peptide precursor ions. These z-ions are then subjected to HCD fragmentation, which produces w-ions of different masses, depending on whether the residues contain leucine or isoleucine. This combined fragmentation method, used in e.g., EThcD-based workflows, has been shown to reliably distinguish between these two isomers, allowing to accurately quantify the Leu- and/or Ile-containing peptides.30
Data acquisition and evaluation
Optimal expertimental setup is determined by several critical factors, including:
- Analysis type (qualitative or quantitative),
- Nature of the sample (a single protein with known mass or a complex mixture of unknown composition),
- Level of analysis (peptides or intact proteins).
Each setup requires tailored analytical strategy and selection of appropriate parameters, which can influence method efficiency and reliability of the results. Choosing the right data acquisition mode, such as data-dependent acquisition (DDA) or data-independent acquisition (DIA), along with analytical parameters like collision energy, can be particularly challenging. These settings must be carefully adjusted to the specific research objective. DIA is often considered a preferred method for analysing host cell proteins (HCPs), owing to its ability to detect diverse and low-abundance protein species. In contrast, targeted analysis of specfic proteins requires a more focused approach, such as targeted MS/MS or parallel reaction monitoring (PRM).
Last but not least is the issue of correct interpretation of the results. A single MS/MS run generates a vast amount of data, requiring specialized software for processing and preliminary analysis. While these tools are extremely helpful, there are interpretation issues that software alone cannot resolve. This is where the expertise and critical thinking of an analyst matter the most. One example of such a situation is when two or more of analyzed peptides have identical or nearly identical masses, making their identification problematic without a careful inspection of fragmentation patterns. In some cases, software may attempt to explain unexpected mass shifts by automatically assigning modifications, that do not actually exist.
Finally, it is important to be aware that public databases are not always reliable. Errors in protein sequences do occur, as confirmed by our own observations. For this reason, the reference sequence should always be cross-checked against other trustworthy sources, such as patent literature, to ensure validity of the results.
Advanced analytical strategies for peptide and protein characterization (MS integration with various separation techniques)
The characterization and quality control of therapeutic proteins require cutting-edge analytical strategies that combine sensitivity, selectivity, and structural insight. Although mass spectrometry is a powerful tool on its own, its performance can be markedly amplified by integration with orthogonal separation methods, including nanoLC, CE, 2D-LC, sample fractionation, and Ion Mobility Spectrometry (IMS). These strategies, examined below, enhance analytical depth and ensure maximum confidence in data quality and regulatory compliance.
Two-dimensional liquid chromatography coupled with mass spectrometry (2D-LC-MS) is an advanced analytical platform that integrates two orthogonal chromatographic separations with high-resolution mass detection. This multidimensional approach enhances separation capacity, improves resolution of structurally similar species, and enables in-depth structural characterization of complex biomolecules such as therapeutic proteins. The method is particularly useful when a single chromatographic technique fails to resolve all analytes or when initial separation is performed under conditions not compatible with MS detection.
One of the most common applications of 2D-LC-MS, which showcases its excellent performance, is characterization of structurally heterogeneous biomolecules, such as recombinant monoclonal antibodies (mAbs). Due to the post-translational modifications, monoclonal antibodies may come with many different variants with distinct physicochemical properties (e.g., size and charge), which are difficult to resolve with traditional one-dimensional methods. Moreover, the analytical workflow is frequently complicated by the incompatibility of mobile phases used in the first chromatographic dimension, such as ion-exchange (IEX) or size-exclusion chromatography (SEC) with mass spectrometric detection. These techniques rely on non-volatile, salt-rich buffers that can suppress ionization efficiency in electrospray ionization (ESI) and contaminate the MS source. To address this limitation, innovative 2D-LC-MS platforms have been developed in which analytes are first separated using MS-incompatible mobile phases, and then transferred to a second dimension (typically reversed-phase chromatography) using MS-compatible solvents, which enable MS detection.31
Ion Mobility Mass Spectrometry (IM-MS) is a gas-phase electrophoretic technique that enables the separation of analytes within a buffer gas based on their charge (z), mass (m), and mobility (K). The mobility parameter K is related to the rotationally averaged collision cross section (CCS) of the ions, reflecting their size and shape in the gaseous phase. The IM-MS method involves measuring the drift velocity of ions as they travel through a buffer gas-filled region under the influence of an electric field. Originally introduced in the 1960s, IM-MS has evolved into a versatile analytical platform and is now routinely coupled with various mass spectrometry (MS) systems. Depending on the configuration, mass analysis can be performed either before or after ion mobility separation. Regardless of the specific setup, all IM-MS systems share key operational principles. Ions are first generated in an ionization region and then directed into the drift region by ion optics and gas flow. Ion pulses are introduced into a drift cell containing buffer gas at a known pressure. An electric field applied across the cell drives the ions forward, while collisions with the buffer gas slow their motion. Importantly, larger ions, which have a greater collision cross-section, experience more frequent interactions with gas molecules, causing them to drift more slowly than smaller ions. This differential mobility allows for effective separation based on molecular size and conformation.32
Ion mobility mass spectrometry (IM-MS) introduces a new level of resolution to biopharmaceutical analysis. This hybrid technique excels at identifying critical quality attributes, including glycosylation patterns, post-translational modifications, aggregation, and quaternary structure changes. Native-mode IM-MS effectively separates conformers and co-eluting species, providing a clearer view of protein structure and dynamics. In the case of antibody–drug conjugates (ADCs), it enables precise determination of drug-to-antibody ratio (DAR) values.33 IM-MS has been also successfully applied to the analysis of protein and peptide aggregates, enabling their structural characterization and even real-time monitoring of formation and molecular dynamics.34
The characterization of biopharmaceutics, including therapeutic proteins and peptides, requires analytical approaches that ensure high sensitivity, low sample consumption, and ability to resolve complex molecular mixtures. Two prominent techniques that meet these criteria are nanoflow liquid chromatography–mass spectrometry (nanoLC-MS) and capillary electrophoresis–mass spectrometry (CE-MS). Both of these platforms have repeatedly proven their value in structural and functional analysis of therapeutic proteins. NanoLC-MS is essential in proteomic workflows due to its high-throughput capability and exceptional analytical sensitivity, achieved with the use of low flow rates (<1 µL/min) which determine efficient nano-electrospray ionization (nanoESI). CE-MS, on the other hand, is a particularly effective method for charge variant analysis (CVA).35
Summary
Mass spectrometry is a powerful and versatile analytical technique for structural analysis of biopharmaceutical products. As pointed out in the FDA’s comprehensive review of BLAs submitted in 2000-2015 period, MS-based methods have been integral to the characterization of licensed therapeutic proteins and associated impurities. Of 80 analysed BLAs, 79 were found to utilize various MS workflows.4 The FDA’s report also demonstrated that the use of mass spectrometry has become increasingly prevalent over the analyzed period, slowly replacing traditional methods. The scope of MS application is also expanding, encompassing a growing number of quality attributes. Given its key role, the vast majority of biopharmaceutical companies, both large and small, incorporate mass spectrometers into their laboratories. However, several conditions must be met before high-quality mass spectra can be generated, obtaining precise and accurate results. One of them is careful selection of appropriate mass spectrometer that aligns with specific analytical objectives and regulatory requirements. Other critical factors that determine the quality of the obtained results include sample preparation, data acquisition methods, and software setup. The optimization process is extremely complex and laborious; however, when properly handled, MS method can provide high-quality data that are difficult, if not impossible, to obtain using other analytical methods.
Prepared by:

Małgorzata Urbaniak
Senior Specialist in Physicochemical Analytical Methods
References
- Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T. Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2(8):151-3.
- Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10 000 daltons. Anal Chem. 1988;60(20):2299-2301.
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246(4926):64-71.
- Rogstad S, Asmelash B, Sommers C, et al. A retrospective evaluation of the use of mass spectrometry in FDA biologics license applications. J Am Soc Mass Spectrom. 2017;28(5):786-94.
- Rogstad S, Mans J, Oyugi M, Asmelash B, Sommers C. The use of mass spectrometry in therapeutic protein biologics license applications: a retrospective review revisited. J Am Soc Mass Spectrom. 2023;34(10):1867-76.
- Hemling ME. Fast atom bombardment mass spectrometry and its application to the analysis of some peptides and proteins. Pharm Res. 1987 Feb;4(1):5–15,
- Wait R. Fast atom bombardment mass spectrometry of peptides. Methods Mol Biol. 1993;17:237–83.
- Winkler HU, Beckey HD. Field desorption mass spectrometry of peptides. Biochem Biophys Res Commun. 1972 Jan 31;46(2):391–8.
- Neagu AN, Jayathirtha M, Baxter E, Donnelly M, Petre BA, Darie CC. Applications of tandem mass spectrometry (MS/MS) in protein analysis for biomedical research. Molecules. 2022 Apr 8;27(8):2411.
- Delobel A, Mass Spectrometry of Glycoproteins: Methods and Protocols. Methods in Molecular Biology, vol 2271. New York (NY): Humana Press; 2021.
- Banerjee S, Mazumdar S. Electrospray ionization mass spectrometry: a technique to access the information beyond the molecular weight of the analyte. Int J Anal Chem. 2012;2012:282574.
- Nishikaze T, Takayama M. Study of factors governing negative molecular ion yields of amino acids and peptides in FAB, MALDI and ESI mass spectrometry. Int J Mass Spectrom. 2007 Nov 15;268(1):47–59.
- Wells JM, McLuckey SA. Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 2005;402:148–85.
- Han X, Aslanian A, Yates JR 3rd. Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008 Oct;12(5):483–90.
- Mikesh LM, Ueberheide B, Chi A, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2007 Jan;1764(12):1811–22
- Waddell Smith R. Mass spectrometry. In: Siegel JA, Saukko PJ, Knupfer GC, editors. Encyclopedia of forensic sciences. 2nd ed. San Diego: Academic Press; 2013. p. 603–8.
- Zubarev RA, Makarov A. Orbitrap mass spectrometry. Anal Chem. 2013;85(11):5288–96.
- Jiang Y, Rex DAB, Schuster D, Neely BA, Rosano GL, Volkmar N, et al. Comprehensive overview of bottom-up proteomics using mass spectrometry. ACS Meas Sci Au. 2024 Jun 4;4(4).
- European Medicines Agency. Guideline on similar biological medicinal products. London: European Medicines Agency; 2014. Report No.: EMA/CHMP/BWP/247713/2012.
- FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: U.S. Food and Drug Administration; 2015.
- Wang Y, Li X, Liu YH, Richardson D, Li H, Shameem M, Yang X. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. MAbs. 2016 Nov-Dec;8(8):1477-86
- Delobel A. Glycosylation of therapeutic proteins: a critical quality attribute. Methods Mol Biol. 2021;2271:1-21.
- Matsumoto H, Komori N, Haniu H. Determination of protein molecular weights on SDS-PAGE. Methods Mol Biol. 2019;1855:101-5.
- Kafader JO, Melani RD, Schachner LF, Ives AN, Patrie SM, Kelleher NL, et al. Native vs denatured: an in-depth investigation of charge state and isotope distributions. J Am Soc Mass Spectrom. 2020 Mar 4;31(3):574–81.
- Duncan MW, Gibson DS. Mass spectrometry for peptide and protein analysis. In: Peptides and proteins
- Po A, Eyers CE. Top-down proteomics and the challenges of true proteoform characterization. J Proteome Res. 2023 Nov 8;22(12):3663–75.
- He L, Weisbrod CR, Marshall AG. Protein de novo sequencing by top-down and middle-down MS/MS: limitations imposed by mass measurement accuracy and gaps in sequence coverage. Int J Mass Spectrom. 2018 Apr;427:107–13.
- Larger PJ, Breda M, Fraier D, Hughes H, James CA. Ion-suppression effects in liquid chromatography–tandem mass spectrometry due to a formulation agent: a case study in drug discovery bioanalysis. J Pharm Biomed Anal. 2005;39(1):206–16.
- Jiang P, Li F, Ding J. Development of an efficient LC-MS peptide mapping method using accelerated sample preparation for monoclonal antibodies. J Chromatogr B Analyt Technol Biomed Life Sci. 2020 Jan 15;1137:121895.
- Zhokhov SS, Kovalyov SV, Samgina TY, Lebedev AT. An EThcD-based method for discrimination of leucine and isoleucine residues in tryptic peptides. J Am Soc Mass Spectrom. 2017 Aug;28(8):1600–9.
- Camperi J, Goyon A, Guillarme D, Zhang K, Stella C. Multi-dimensional LC-MS: the next generation characterization of antibody-based therapeutics by unified online bottom-up, middle-up and intact approaches. Analyst. 2021;146(3):747–69.
- Harvey SR, MacPhee CE, Barran PE. Ion mobility mass spectrometry for peptide analysis. Methods. 2011 Aug;54(4):454–61.
- Skeene K, Khatri K, Soloviev Z, Lapthorn C. Current status and future prospects for ion-mobility mass spectrometry in the biopharmaceutical industry. Biochim Biophys Acta Proteins Proteom. 2021 Dec;1869(12):140697.
- Depraz Depland A, Stroganova I, Wootton CA, Rijs AM. Developments in trapped ion mobility mass spectrometry to probe the early stages of peptide aggregation. J Am Soc Mass Spectrom. 2023 Feb;34(2):193–204.
- Lechner A, Giorgetti J, Gahoual R, Beck A, Leize-Wagner E, François YN. Insights from capillary electrophoresis approaches for characterization of monoclonal antibodies and antibody drug conjugates in the period 2016–2018. J Chromatogr B Analyt Technol Biomed Life Sci. 2019 Aug 1;1122–1123:1–17.
Related resources
Rapid Microbiological Methods in Quality Control of Sterile Drugs
Contamination, Drug product, Microbiology

From Research to Release: Mapping Mabion’s Full Analytical Lifecycle for Monoclonal Antibodies
Analytics, Mabion, Monoclonal antibody

The New Era of Biosimilar Development: Seizing the Opportunity Under EMA’s Streamlined Guidelines
Biosimilars, Clinical trials, Drug development, EMA, FDA, Mabion, Regulatory