Co-Development Partnership Models in Biologics Programs
- Co-development partnership in biologics differs from traditional fee-for-service outsourcing by aligning the CDMO’s success with the sponsor’s outcomes. The result is a jointly invested effort where both parties stand to benefit from a biologic’s success, fostering deeper collaboration than transactional contracts ever could.
- Early-stage engagement is crucial for maximizing the value of co-development. Co-development partnership initiated in these early phases ensure that process robustness and quality are “baked in” from the start, avoiding expensive rework or comparability challenges down the line.
- Co-development has particular resonance in rare disease and orphan drug development, where traditional models struggle. These projects carry high scientific risk and small patient populations, deterring many service providers.
Co-Development Partnership as a Value-Driven Model
Co-development indicates a fundamental shift in the collaboration pattern between biopharma and CDMOs. Traditionally, a CDMO (Contract Development and Manufacturing Organization) was a hired contractor: paid a fee to perform discrete tasks, with an eye on minimizing its own costs and risks. Often this meant a narrow focus on meeting minimum requirements cheaply. After fulfilling their contractual obligations, many profit-driven vendors reduced their support for the project in order to minimize costs, which frustrated biotech innovators who found their programs languishing due to inadequate effort. The incentives were misaligned. In such a situation, finding a trusted CDMO that will be a partner and provide real support could become a decisive factor for the success of the project. Co-development flips this script. In a co-development partnership, the CDMO becomes a stakeholder in the program’s success, not just a contractor. Both parties commit resources to co-create the drug’s value. Rather than trying to minimize their involvement, the CDMO deliberately increases its risk exposure, for example by deferring certain fees in return for a future payoff if the drug succeeds. This shared-risk model “marks a significant departure from the traditional fee-for-service approach”.
The immediate effect is to align incentives: the CDMO only wins if the drug wins. Quality and efficiency thus become paramount for everyone. Cutting corners is no longer tenable when the partner has skin in the game. Instead, the CDMO’s deep expertise is fully leveraged to solve problems and make the project a success, since their own reward (milestone payments, royalties, equity in the product, etc.) depends on it. For the sponsoring company, this arrangement reduces upfront costs and financial burden, essentially giving them more chances to advance viable drug candidates with limited capital.
Importantly, this value-driven model doesn’t just feel good on paper. It can materially improve outcomes. When a CDMO steps up as a co-investor, it brings to bear its full arsenal of capabilities (seasoned scientists, specialized infrastructure, project management rigor) in a proactive way. Meanwhile, the biotech sponsor is more willing to trust and share information openly, knowing their partner is equally vested. The result is often a more efficient development process. Empirically, drugs developed through alliances and co-development have shown higher success rates in clinical trials compared to those developed in independently.1 By pooling expertise and aligning goals, co-development partnership create a synergistic effect. This value-centric approach is setting a new benchmark in the industry, “elevating the status of CDMOs from mere vendors to key innovation partners” who actively contribute ideas and intellectual property to the program. For many biopharma companies, especially resource-strapped start-ups, a co-development deal with a capable CDMO it’s a strategic alliance to maximize the drug’s potential. Mabion announced the implementation of this approach in our business strategy, going beyond the scope of an exclusive service agreement alone.
Preclinical and Early Clinical Phases as the Preferred Window for Co-Development Partnership
Timing is everything when it comes to entering a co-development partnership. The preclinical and Phase 1 clinical stages emerge as the sweet spot for such collaborations.
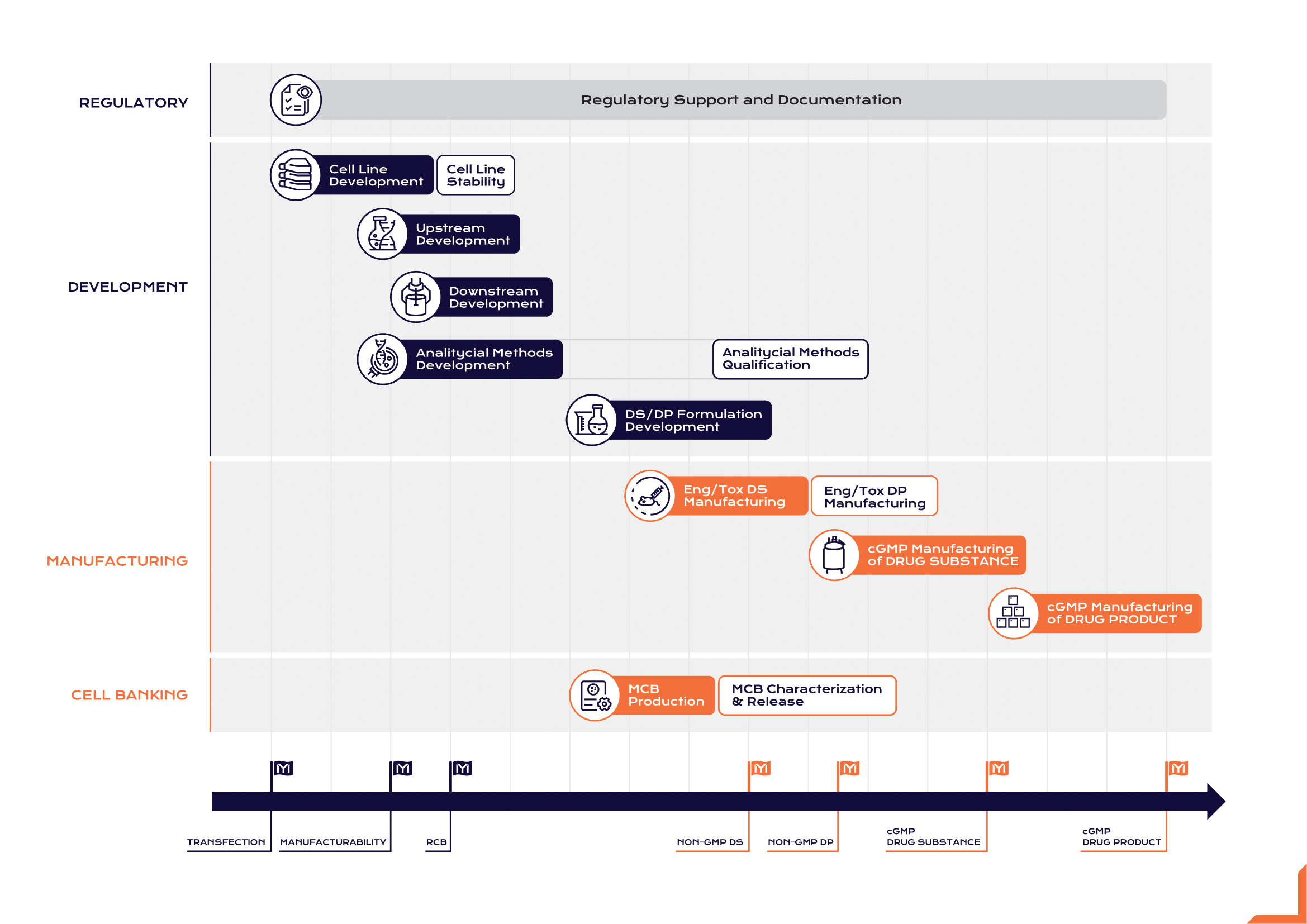
By preclinical (late research through IND-enabling studies), a drug candidate has shown enough promise biologically to merit serious development, but it still faces a long road of uncertainty ahead. This is exactly where a dedicated CDMO partner can make the biggest impact and where the sponsor most needs help. In preclinical development of a biologic, crucial groundwork is laid: cell line development, process development for upstream and downstream, formulation optimization, analytical method development, and pilot production of material for animal studies. Decisions made here will echo throughout the drug’s lifecycle.
Process matters immensely for biologics. Small changes in manufacturing conditions or formulation can ripple into significant differences in the final biotherapeutic’s quality, safety, or efficacy. After a certain point, you cannot easily change course without going back to regulators to prove comparability.
Imagine trying to implement a better purification method or a new cell line after you’ve dosed patients in Phase 3. The regulators would demand extensive bridging studies to ensure the “new” product is equivalent to the old. That’s a costly, time-consuming detour (if it’s even feasible). Therefore, locking in an optimal process early is paramount.
By engaging in co-development during preclinical or Phase 1, sponsors ensure that the process and product profile are optimized from the start, with the CDMO’s experts guiding development to avoid pitfalls that might derail the drug later.
From a CDMO perspective, the preclinical to Phase I window is also the preferred entry point. At this stage, the number of viable drug candidates has been winnowed to those with real potential. But the technical risk remains high. Many candidates will still fail due to toxicity, lack of efficacy, or other issues in early human trials. Traditional CDMOs might shy away from taking on risk here, but under co-development terms, the CDMO shares the risk knowingly. It’s worth noting that engaging too early (at the very basic research or discovery stage) is often impractical because the attrition rate of early targets is exceptionally high (only a tiny fraction ever make it to clinical testing).
It’s a calculated gamble: by contributing know-how and resources in early development, the CDMO can help steer the program onto a strong trajectory, increasing its chances of success. In fact, establishing a robust, scalable manufacturing process and demonstrating early manufacturability can itself de-risk a program significantly. Studies have shown that when development is done in alliance with an experienced partner, later-phase success probabilities improve.
Early co-development involvement helps sponsors learn sooner rather than later whether a biologic is developable, and if it is, sets it up to smoothly advance into Phase 2 and 3. The CDMO ensures that cell culture yields are sufficient, the purification process consistently achieves high purity and removes impurities, and the formulation keeps the biologic stable – all by the time Phase 1 trials are underway.
Notably, Phase 1 clinical trials represent a last practical entry point for co-development in many cases. By Phase 2, the die is largely cast in terms of the drug’s process and formulation. Changing your manufacturing paradigm mid-stream (for instance, switching to a new production cell line or a radically different process) could invalidate earlier clinical data or require extensive comparability testing.
Moreover, by Phase 2 or 3, the sponsor might have already invested heavily and proven the concept, which typically makes them less inclined to give away a share of the asset in a partnership or conversely, the asset becomes so valuable that a CDMO’s cost-sharing offer is less attractive relative to traditional financing.2
Thus, the best window for co-development is from preclinical up to Phase 1 (and sometimes into Phase 2a for certain projects). At these stages, the alignment is perfect:
- the sponsor needs the CDMO’s comprehensive capabilities to get through the perilous early development quickly and effectively
- the CDMO is willing to bet on the program’s success, because its expertise can substantially influence the outcome.
Together, they craft the development plan, de-risk CMC (Chemistry, Manufacturing and Controls) aspects early, and set up the drug for a smoother clinical journey. By the time the program reaches a critical inflection (say, Phase 2 proof-of-concept), both partners have significantly de-risked the technical aspects and stand to gain from the progress. Co-development partnership thrives when there is a defined candidate and a clear development path, but still enough unknowns that the CDMO’s contributions will make a big difference. Preclinical and Phase 1 hit that sweet spot.
Integrated Co-Development Capabilities Supporting Drug Development at Mabion
Mabion’s co-development offering is built on integrated, end-to-end capabilities that support virtually every aspect of biologic drug development. Rather than providing a la carte services in silos, Mabion offers to walk side by side with its partners from Gene to Vial. This includes cell line development, process development (upstream and downstream), analytical method development and validation, drug substance manufacturing, and regulatory support. With the support of its strategic partners, Mabion can seamlessly coordinate the complex puzzle of biologics R&D.
For a co-development partner, that means fewer hand-offs and no cracks for the project to fall into. The manufacturing process and quality controls co-evolve with the molecule’s development, guided by a single experienced team. Mabion’s approach exemplifies how a CDMO can act as the engine of development for a small biotech’s novel biologic, while the biotech focuses on the drug’s discovery and clinical strategy.
Mabion has also distinguished itself with experience in orphan drugs and biologics for rare diseases, an area many larger players hesitate to venture. These projects are often high-risk, high-reward: scientifically challenging, with tiny patient populations and unclear commercial returns. Yet, Mabion has demonstrated commitment to such programs, driven by the ethos that every life is worth saving. An ethos many of Mabion’s partners share. In practice, we implemented our own initiatives concerning orphan drugs. We have obtained FDA Orphan Drug Designation status for our monoclonal antibody (Mabion CD20; rituximab biosimilar) in two rare indications (membranous nephropathy and autoimmune hemolytic anemia). This not only underscores Mabion’s expertise with biologics in niche indications, but also signals to potential partners that Mabion understands the regulatory and development nuances of orphan therapies.
Orphan and rare disease biologics are drugs that, without a partnership model, might never be made at all. The patient populations are too small, the commercial prospects too uncertain for a single company to bear. By sharing the risk, Mabion and its partners ensure that no promising therapy is left unexplored solely due to financial risk. Every project is a gamble, but when successful, the reward is measured in lives improved or saved. For a rare disease patient and their family, the impact of such a partnership is enormous: it can mean the difference between no treatment and a new hope.3
Value-Sharing Frameworks in Biologics Co-Development Partnership
At the heart of the co-development concept lies a simple but powerful principle: shared investment for shared reward. In practical terms, this means designing a risk- and value-sharing framework for each partnership. Instead of the sponsor paying a flat fee regardless of outcome, the co-development contract ties the CDMO’s compensation to the drug’s success in some way. There are various models for this:
- One common approach is for both parties to split the costs of development (for example, each pays 50% of the process development and clinical manufacturing expenses) and, in exchange, the CDMO will receive a predetermined share in the product’s future revenues (royalties or profit split) if and when it reaches the market.
- Another approach might involve the CDMO giving the sponsor discounted rates or credit for services upfront, with the difference recouped as milestones: if the drug achieves certain milestones (e.g. successful Phase 2, or BLA approval), the CDMO gets bonus payments or equity that reflect the value created.
- More novel structures include cases where the CDMO actually co-founds a new venture around the product, taking an equity stake in the biotech in lieu of some fees, or where a CDMO might license intellectual property it developed (such as a cell line or a formulation technology) to the program in return for a downstream payment.
The immediate benefit of risk-sharing frameworks is that they lower the financial barriers for the sponsor. Small biotechs often operate on limited budgets (seed or Series A funding) that make the high cost of biologics development daunting. A risk-sharing deal can, for instance, cut the upfront cash outlay needed to get through Phase 1 in half, or more, compared to paying a traditional CDMO in full. This can literally make the difference between a program moving forward or being shelved.
For the CDMO, of course, this means taking on greater risk – if the drug fails, they will not recoup those deferred costs – but in return they stand to earn much more than the typical service fee if the drug succeeds. It’s a high-risk, high-reward business model for the CDMO as well.

The contract often encodes this spirit: for example, outlining decision-making structures like joint steering committees, or defining how unexpected costs will be handled collaboratively rather than just passed along to the sponsor. In essence, the framework creates a single, combined team from two organizations.
One tangible outcome of these frameworks is a more efficient development process. When cost is not simply a burden on one party, there is more freedom to do what’s scientifically or technically right for the product. Need an extra optimization experiment? Under pure fee-for-service, the sponsor might hesitate due to budget, or the CDMO might not bother if it’s outside scope; in a value-sharing setup, both know that investing in a better process now will pay off later for everyone, so there’s far more willingness to go the extra mile. This often leads to higher quality outcomes – better yields, more robust data, fewer regulatory issues – precisely because the team wasn’t cutting corners to save one side money.
Leveraging Complementary Expertise – Shared Risk, Shared Reward
Co-development in biologics is as much about brains as it is about money. A critical advantage of these partnerships is the ability to leverage complementary expertise from each party, creating a synergy that can drive a project faster and smarter through the pipeline. Biotech innovators and academic spin-offs often bring groundbreaking science. They have deep insight into the disease and the mechanism of action, and often a patent or proprietary technology. What they may lack are the practical development capabilities: process engineering, biomanufacturing scale-up, quality control systems, clinical trial material supply chain, and regulatory strategy. On the flip side, CDMOs specialize in precisely those areas. They’ve run dozens of cell culture processes, purified proteins of all stripes, navigated countless GMP audits, and filed CMC sections for many biologics.
When these two sides join forces under a co-development model, the resulting team is far more formidable than either alone. The biotech’s scientists work hand-in-hand with the CDMO’s process development engineers, each learning from the other. The knowledge transfer is bidirectional. In effect, the biotech gains a fully-fledged development department overnight, and the CDMO becomes an extension of the biotech’s R&D team, but with the benefit of having seen many molecules before. The combined brainpower and experience can be the decisive factor that gets a complex biologic to the finish line.4
It’s also worth pointing out that these frameworks help spread risk in a portfolio sense. A CDMO might engage in a handful of co-development projects, knowing some will fail and some may succeed wildly. The returns from the winners can offset the losers, much like a venture capital model. The sponsoring biotech, on the other hand, essentially gains a form of project insurance. They are not alone bearing the full brunt if the program fails, which can be reassuring to their investors and stakeholders.
This shared burden can enable more projects to move forward in the ecosystem. From a big-picture view, risk-sharing partnerships can thus increase the overall R&D productivity of the biopharma industry. More shots on goal, more efficient use of capital, and potentially a higher success rate per project started are reasons such models are gaining traction, particularly in biologics where the stakes (and costs) are high.
Regulatory Approval, and Commercial Supply Chain Execution of Co-Developed Biologics
A biologic’s journey doesn’t end when clinical trials succeed. In fact, some of the most critical challenges lie in regulatory approval and commercial manufacturing, where co-development partnership continue to prove their value. Bringing a biologic to market requires navigating a labyrinth of regulations and then scaling up production to supply potentially tens of thousands of patients. In a co-development model, the partners tackle these challenges side by side, which greatly smooths the path compared to a hand-off approach.
On the regulatory front, a strong CDMO partner can significantly streamline the preparation of approval submissions. Biologics require voluminous documentation for regulatory review – particularly the CMC section of a Biologics License Application (BLA) in the US or the Marketing Authorization Application (MAA) in Europe. Regulators demand precise data demonstrating that the manufacturing process is well-controlled, the product is consistently made to high quality, and all analytical methods are validated. A co-development CDMO, having been involved since preclinical or early clinical stages, possesses intimate knowledge of the product and process. It has been collecting the necessary data (stability studies, batch analyses, validation protocols) all along, with an eye toward the final submission. There’s no scramble to retrofit quality systems or generate missing studies at the last minute. The groundwork is integrated into the development plan. Moreover, an experienced CDMO is adept at regulatory strategy. It understands the differences between EMA and FDA requirements, and can ensure the dossier is prepared to satisfy both, avoiding delays or refusals.
The key advantage is continuity. The same team that helped develop the drug now helps defend it to regulators, lending credibility. This not only increases the chance of a first-cycle approval, but also can make the approval faster. It’s not uncommon for CDMOs to have prior experience getting similar products approved, which they can leverage to prevent common pitfalls. Once regulatory green lights are obtained, the focus shifts to commercial execution. This is where the earlier investments in process robustness pay off.
New challenges emerge, like scaling up manufacturing volume, managing supply logistics, and maintaining cost-efficiency. In a traditional scenario, a biotech that developed a drug might now have to find a manufacturing partner for commercialization or invest in building its own facility, both of which are huge undertakings. In the co-development scenario, however, the transition to commercial production is often pre-negotiated and seamless: the CDMO partner typically continues as the commercial manufacturer (often with a long-term supply agreement that was part of the co-development deal). This continuity is essential. The process used in Phase 3 can be directly scaled up or tech-transferred within the CDMO’s facilities for larger bioreactors or production lines. Because the CDMO was involved in development, there is no knowledge gap to overcome in scale-up. Well-prepared CDMO know where the risks are and can handle the scale increase efficiently.5
Quality assurance and compliance remain front and center during commercial manufacturing. With both parties invested, there’s no skimping on GMP compliance. The co-development CDMO works continuously to ensure that production meets the rigorous standards for commercial distribution. This includes regular quality audits (often the partner will have rights to audit the CDMO and vice versa), ongoing process verification, and updates to regulatory filings for any post-approval changes. Because the regulatory environment can evolve, having a partner who stays on top of compliance trends means the product stays in compliance throughout its life cycle. Additionally, a co-development partnership often contemplates market expansion. When the drug needs approval in more countries, the CDMO helps generate whatever additional data or documentation might be required.
Another aspect is commercial supply chain management. Both partners should ensure that, once the drug is on the market, patients will be able to obtain it reliably. The CDMO’s operations team will manage the scheduling of manufacturing campaigns, stockpiling of inventory, and scaling of capacity as demand grows. In a risk-sharing model, sometimes the CDMO even agrees to maintain a certain safety stock at its own cost, knowing that its revenue will come from product sales. This buffers any unforeseen spikes in demand or production slowdowns. The co-development partnership might also plan for redundancy.
With biologics, managing the cold chain is vital. All these operational details are handled professionally by the CDMO partner, allowing the sponsoring company to focus on commercial strategy.
Conclusion
In summary, the rise of co-development partnership models in biologics is reshaping the biopharma landscape. This approach turns traditional client-vendor dynamics into true alliances, aligning incentives so that both a biotech innovator and a CDMO partner share the risks and the triumphs of bringing a biologic to market. By engaging before the first patient is ever dosed co-development partners are able to embed quality, scalability, and efficiency into the DNA of a drug’s development process. The benefits of this model are manifold.
Co-development has proven especially valuable in challenging arenas like rare diseases, where the traditional models falter and creative risk-sharing can mean the difference between a therapy reaching patients or dying on the vine. Of course, co-development is not without its demands: it requires trust, transparency, and meticulous structuring to ensure each party is fairly rewarded. But as more success stories emerge the appeal of this model grows. In an era of expanding biologic modalities and intense pressure to innovate, co-development partnership offer a compelling way forward. They exemplify a powerful principle in biopharma today. When partners unite their strengths and share the load, even the most formidable drug development challenges can be overcome – together.
Download our free e-book, which transparently shows how we operate. Choose Mabion Biologics CDMO as your partner!
Prepared by:

Business Development Director

Senior Director of Business Development

Marketing Specialist
References
- Kurata H, Ishino T, Ohshima Y, Yohda M. CDMOs Play a Critical Role in the Biopharmaceutical Ecosystem. Front Bioeng Biotechnol. 2022; 10: 841420.
- Steadman VA. Drug Discovery: Collaborations between Contract Research Organizations and the Pharmaceutical Industry. ACS Med Chem Lett. 2018; 9(7): 581-583.
- Debnath A, Mazumder R, Mazumder A, Tyagi PK, Singh RK. Challenges and Progress of Orphan Drug Development for Rare Diseases. Curr Pharm Biotechnol. 2025.
- Djurian A, Makino T, Lim Y, Sengoku S, Kodama K. Dynamic Collaborations for the Development of Immune Checkpoint Blockade Agents. J Pers Med. 2021; 11(6): 460.
- Danzon PM, Nicholson S, Pereira NS. Productivity in pharmaceutical-biotechnology R&D: the role of experience and alliances. J Health Econ. 2005; 24(2): 317-339.