Strategies for High-Quality Recombinant Protein Production
- Recombinant protein production is traditionally undertaken in mammalian cell culture. The drug substance is an ensemble of closely related molecular species (glycoforms, charge and size variants), so control must target populations, not a single structure.
- Across recombinant modalities quality is an emergent property of a coupled system that includes the expression host, the upstream environment, the downstream purification train, the formulation and container closure, and the analytical ecosystem that measures and governs critical quality attributes (CQAs) across scale and time.
Factors Affecting the Quality of Recombinant Protein Production
Recombinant protein production is most rigorously defined as the consistent achievement of a clinically justified Critical Quality Attribute (CQA) set through a quality control strategy. In biologics the dominant determinants of this CQA landscape originate bioprocessing. Early choices in upstream and bioreactor production mode establish a area for product quality that downstream purification can only bias, not fully redefine. From a process-structure-function perspective, this means that quality is an emergent property of the coupled host–process system.
Lot-to-lot differences in media, supplements, resins, and filters can shift cell metabolism and impurity clearance kinetics in recombinant protein production. Proteins analytical characterization must be orthogonal, because attributes that are not measurable with adequate sensitivity cannot be reliably controlled or trended. Lifecycle analytical method management are integral to sustaining control during recombinant protein scale-up and technology transfer, where comparability arguments depend on reproducible measurement of subtle distributions rather than single-point metrics.1
Selecting Expression Systems for Recombinant Protein Production
Within mammalian expression systems, CHO remains the practical gold standard for commercial recombinant protein production. It combines high achievable titers with robust suspension growth in chemically defined media and a long regulatory history that reduces development and approval friction. Historical regulatory approvals have established CHO-based systems dominate approved mAb manufacturing, reinforcing its platform status and the ecosystem of tools that support its use at industrial scale.
In our CDMO experience, the strategic value of CHO in recombinant protein production is not only its productivity but also the predictability of its manufacturability when paired with a disciplined cell line development workflow that controls genetic stability, secretion competence, and post-translational modification (PTM) machinery within a defined process envelope. That predictability matters because expression host selection is a decision with asymmetric risk. It is relatively easy to change media or chromatography resins later, but it is difficult to change the cell substrate late in development without triggering extensive comparability work, and for biosimilars the host and clone strategy must also support fine-grained matching of glycosylation and variant profiles to the reference product.
The choice among CHO cell lines and engineered derivatives can materially change upstream performance and product quality. Common lineages include CHO-K1, CHO-S, and DHFR-deficient variants such as CHO-DG44 and CHO-DXB11, each with differences in metabolic behavior, stress responses, and suitability for particular selection systems.2 Clonal variation is a central reason why CHO can be both powerful and challenging, because different clones, even within the same lineage, can exhibit different N- and O-glycosylation profiles, protein processing capacities, and responses to media composition and feeding strategies.3
DHFR-based amplification approaches are often associated with CHO-DG44 backgrounds, while other platforms use glutamine synthetase (GS) selection and amplification strategies, and both selection architectures influence the distribution of expression levels, the time to stable clone generation, and the risk of copy number instability. These are critical factors in recombinant protein production, as the selection strategy shapes the population from which high-producing clones are isolated and therefore can indirectly influence quality through clonal heterogeneity in PTM pathways, secretion stress, and protease release profiles. From the perspective of manufacturability, we treat clone selection as a multi-objective optimization problem.4
CHO engineering uses tools such as CRISPR-Cas systems to modulate metabolic flux, apoptosis pathways, and glycosylation enzymes.5 That reported outcomes including improved culture longevity, higher titers for multiple antibodies, or targeted glycoform changes such as defucosylation to enhance antibody-dependent cellular cytotoxicity (ADCC). For example:
- FUT8 knockout strategies can drive defucosylation and thus change Fc receptor binding and ADCC, but they may also change cellular metabolism and secretory pathway loading, which could indirectly affect aggregation or charge variants, necessitating an expanded analytical panel during development.6
- Clones engineered for anti-apoptotic phenotypes can extend viable culture duration, which can increase cumulative titer but may also increase exposure to proteases and to reactive species that can drive clipping or oxidation unless harvest timing and culture conditions are tightly managed.7
- CHO’s intrinsic glycosylation tendencies also matter. CHO cells can produce glycoforms close to human serum IgG but have limitations such as the lack of certain human sialyltransferase activities, which is why glycoengineering is often applied when higher sialylation or altered galactosylation is desired. Host-driven glycosylation patterns are then modulated by upstream culture conditions.
These strategies can be valuable when the product’s mechanism of action depends on effector function, or when a biosimilar must match a reference product glycan distribution within tight comparability windows.
Process Design Choices That Shape Recombinant Protein Quality
Process design is the bridge between a genetically capable cell factory and a reproducible product quality profile, and in our experience the most successful programs treat upstream and downstream development as a integrated system. Within upstream processing, the fundamental quality levers include medium and feed composition, feeding strategy, and other bioreactor parameters. Upstream design begins with a decision about bioprocess operation mode and scale trajectory. It also includes construct- and secretion-focused interventions that can reduce heterogeneity by improving folding and assembly, which in turn reduces downstream burden.
Industry case examples have shown that removal of hydrophobic regions can enhance secretion of extracellular domains. Rational construct redesign can improve yield and purity for complex, multi-domain proteins. That illustrating how bioprocess design can include molecular-level changes that reshape the manufacturability landscape. In difficult-to-express formats, supplementation with chaperone-like companions has been described as a means to facilitate correct assembly or processing. Such approaches must be assessed for GMP compatibility and impurity risk, they illustrate the broader point that folding and assembly are upstream quality determinants.8
Downstream process development includes more accurately a sequence of separations and transformations that must remove impurities while preserving the product’s native structure and controlling product-related variants in recombinant protein production. Platform purification commonly begins with clarification and viral inactivation (by depth filtration and/or centrifugation) followed by Protein A capture, a second low-pH viral inactivation step, and a polishing sequence using ion exchange (IEX) and/or hydrophobic interaction chromatography (HIC) or mixed-mode resins, followed by ultrafiltration/diafiltration (UF/DF) for concentration and buffer exchange.9
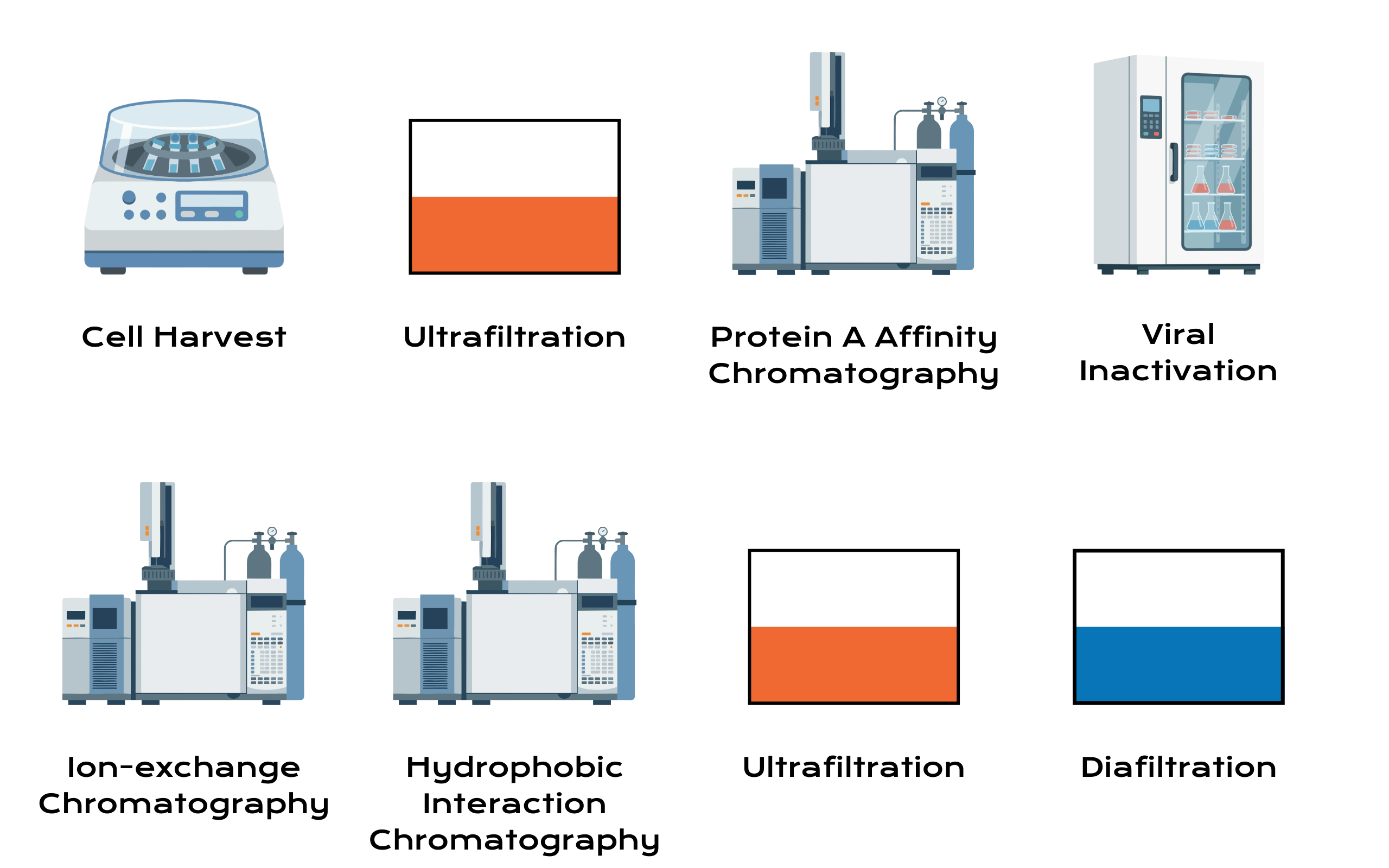
A quality-centric downstream strategy begins at harvest with appropriate clarification design. The burden and nature of impurities entering capture influence both resin performance and product stability. High cell density processes can generate higher levels of HCP, DNA, and colloidal material, requiring carefully selected depth filters and potentially staged clarification to protect capture columns from fouling and to reduce bioburden risk. The capture step, while powerful, introduces its own quality considerations, including leached Protein A, potential co-elution of HCP, and the need to control elution pH and conductivity to avoid aggregation or partial unfolding.10
The low-pH viral inactivation step is a regulatory and safety imperative, but it is also a product stress step, and must be optimized to achieve viral inactivation while minimizing aggregation or chemical degradation. In our development programs, we treat viral inactivation conditions as part of the product’s stress map and link them to aggregation propensity measurements, because the safe pH range can differ across molecules.11
Polishing operations are where downstream design becomes highly product-specific, particularly for charge variant control and aggregate removal. This is often where the most meaningful tradeoffs between yield and quality occur. Cation exchange chromatography (CEX) can be used in bind-and-elute or flow-through modes to manage acidic and basic variants, while anion exchange chromatography (AEX) is commonly used in flow-through to clear DNA, endotoxin, and certain HCP species, but the balance among these steps depends on the impurity profile and on which product variants must be minimized. UF/DF and final filtration are sometimes underestimated as quality drivers, yet they can determine aggregation levels, subvisible particle profiles, and even chemical stability.12
Managing Product-Related Variants in Monoclonal Antibodies
Monoclonal antibodies are inherently heterogeneous, and managing product-related variants is therefore a core competence for any organization that seeks to manufacture high-quality mAbs at scale. The most common categories of product-related variants include:
- Size variants (high molecular weight species such as aggregates and low molecular weight species such as fragments) can be driven by partial unfolding, interfacial stress, or chemical modification.
- Charge variants (acidic and basic species driven by PTMs and processing) can be driven by deamidation, sialylation, glycation, C-terminal lysine processing, and isomerization.
- Glycoform variants (differences in glycan composition, occupancy, and structure) reflect host glycosylation pathways and upstream culture environment.
These variants matter because they can influence potency, Fc-mediated effector functions, pharmacokinetics, and immunogenicity risk, and regulators expect a science-based assessment of their clinical relevance, especially in biosimilar contexts where differences relative to the reference product must be justified. From a manufacturing standpoint, variant management is also about process capability in recombinant protein production, because variants that are difficult to control can constrain yield and scale-up or can create batch failure risk due to specification excursions.13
Variant management requires a holistic process view supported by orthogonal analytics: we must measure variants at multiple points, relate them to process conditions, and then select interventions that minimize risk while preserving yield and manufacturability. For mAbs, the most frequent upstream drivers of variant risk include overly long culture duration (which increases residence time for clipping and oxidation), uncontrolled pH or dissolved oxygen excursions (which affect glycosylation and oxidative stress), and high cell lysis rates (which increase HCP and protease burden). Downstream drivers include aggressive low-pH viral inactivation conditions, poorly controlled chromatography elution windows, excessive filtration pace, and hold times at concentrations or temperatures that promote aggregation or chemical modification.
Analytical Approaches Supporting High-Quality Recombinant Proteins
Analytical panel design should mirror the CQA hierarchy: confirm primary structure, quantify heterogeneity (size/charge/glycans), establish higher-order structure (HOS) comparability, demonstrate potency, and quantify process-related impurities with methods suitable for GMP trending and investigations.
Regulatory agencies emphasize comprehensive characterization including primary structure confirmation, glycosylation and PTM profiling, impurity quantification, and bioactivity evaluation. The EMA guideline provide a law backbone for what constitutes adequate characterization and specification setting, while newer ICH analytical guidelines emphasize method development and validation as lifecycle activities. At Mabion, we operationalize these expectations by building analytical panels that align with decision points in development and manufacturing. That platform analytics focus on comparability and risk identification while late-phase QC testing emphasize routine robustness, transferability, and release suitability.
Structural Analytics for Recombinant Protein Production
For structural parameters, we recommend an analytical panel that begins with identity and primary structure confirmation through intact mass analysis and peptide mapping, supported by HOS analysis using circular dichroism, because these methods provide the most direct evidence that the molecule produced matches the intended structure. Peptide mapping, combined with targeted analysis of disulfide bonds and free thiols can identify scrambling or incomplete pairing that may arise from secretion stress or redox imbalance. In development settings where higher-resolution structural insights are needed, hydrogen-deuterium exchange mass spectrometry and native mass spectrometry can provide additional information on conformational dynamics and complex formation, but these tools must be evaluated for their lifecycle robustness if they are to support GMP decision-making.14
Physicochemical Analytics for Recombinant Protein Production
For physicochemical attributes, we recommend a panel that characterizes size heterogeneity, charge heterogeneity, hydrophobicity-related variants, and glycosylation profile. Size variants are typically assessed by size-exclusion chromatography (SEC) for aggregates and fragments, complemented by orthogonal methods such as capillary electrophoresis under denaturing and non-denaturing conditions or analytical ultracentrifugation when deeper resolution is needed for complex association states. Charge variants are commonly profiled by ion exchange chromatography and capillary isoelectric focusing, and these techniques are particularly important for mAbs because PTMs such as deamidation, sialylation, and C-terminal processing can shift charge distributions in ways that correlate with stability and sometimes potency. Hydrophobic interaction chromatography or reversed-phase methods can provide additional resolution for certain variants, and they are often useful in investigational settings to identify subtle changes that might not appear in SEC or IEX.15
Glycosylation profiling is performed through released glycan analysis with fluorescent labeling and chromatography, coupled with mass spectrometry for structural assignment, and complemented by glycopeptide mapping to connect glycoforms to specific sites. Because glycosylation is a distributional attribute, the panel must quantify relative abundances of key glycan families (e.g., high-mannose, afucosylated, galactosylated, sialylated species) and must be sufficiently reproducible to support comparability studies and lot-to-lot trending.16
Biological Analytics for Recombinant Protein Production
For biological activity, the panel must be tailored to the mechanism of action in recombinant protein production. It typically includes target binding assays and, when relevant, Fc-mediated effector function assays that collectively establish potency and functional integrity. Binding to antigen can be assessed by ELISA and by real-time interaction methods such as surface plasmon resonance, which provide affinity and kinetic parameters and can be sensitive to subtle structural changes in the antigen-binding region or to glycosylation effects that alter Fc conformation. For IgG1 molecules where effector functions matter, flow cytometry cell-based assays that quantify ADCC, apoptosis assay, complement-dependent cytotoxicity (CDC), as well as binding to Fc gamma receptors and C1q, are often central to establishing functional comparability and to linking glycosylation attributes such as fucosylation and galactosylation to biological outcomes.17

Bioassays should be designed with an understanding of their variability and their role in the control strategy, because potency assays can be more variable than physicochemical assays and therefore require careful reference standard management and statistical control. In biosimilar programs, these assays are used to confirm functional similarity, which emphasizes that functional outcomes should align with structural and physicochemical similarity rather than compensate for major differences.18
In our practice, we integrate biological activity results with other data through multivariate trending. This approach can identify early drift signals, such as a gradual increase in high-mannose species that corresponds to a measurable change in Fc receptor binding, thereby enabling proactive process correction.
Translating Recombinant Protein Production Strategies into GMP Manufacturing
GMP manufacturing demands both technical robustness and disciplined execution. We frame GMP translation as a project lifecycle activity aligned with ICH Q10, where process performance and product quality monitoring continue after launch and feed into continual improvement. We manage risk consistent with ICH Q9(R1) to prioritize controls and investigations.19,20
The upstream and downstream strategies in recombinant protein production s must be treated as validated unit operations with defined parameter ranges, in-process controls, and acceptance criteria that reflect both process capability and clinical relevance. This requires a structured process characterization phase that builds on development DoE results and scale-down models to identify CPPs and to justify operating ranges, followed by process performance qualification (PPQ) that demonstrates reproducibility at commercial scale.
A key aspect of GMP process transformation is ensuring that upstream process are qualified and controlled in a way that supports reproducible product quality. Upstream variability can propagate into downstream burden and ultimately into batch variability or failures. Cell banking and characterization must follow expectations consistent with ICH Q5D, including identity, purity, stability, and adventitious agent testing, and operational controls must ensure that passage number, thawing, expansion, and inoculation procedures are consistent and traceable. Upstream GMP control also requires tight management of media and feeds, including supplier qualification, lot-to-lot testing for key attributes, and defined preparation and hold-time controls, because mammalian cell culture systems can be sensitive to subtle variations in raw materials.21
When a process uses intensified modes such as perfusion, GMP translation must also address the complexity of continuous operations, including control of steady-state conditions, management of disturbances, and integration of downstream capacity, aligning conceptually with ICH Q13 principles for continuous manufacturing even when the process is not fully continuous across all unit operations.22
Downstream GMP translation is similarly multidimensional. Viral safety demonstration and ongoing assurance are anchored in ICH Q5A(R2), but the practical GMP implication is that viral clearance steps must be robust to reasonable process variation and that unit operation performance must be monitored over time for drift. Chromatography in GMP requires control of column packing, flow distribution, and resin performance, and it requires trending of key indicators such as pressure-flow behavior, capacity, and impurity clearance, because subtle resin changes can manifest as quality shifts over multiple cycles. UF/DF and sterile filtration steps must be validated not only for yield and filter integrity but also for their influence on aggregates and particles. The overall downstream strategy must also incorporate microbial and bioburden controls, cleaning validation, and facility segregation where appropriate, because contamination risk is managed through both process design and quality system discipline.
FAQ
Prepared by:

Marketing Specialist
References
- Jayakrishnan A, Wan Rosli WR, Tahir ARM, Razak FSA, Kee PE, Ng HS, Chew YL, Lee SK, Ramasamy M, Tan CS, Liew KB. Evolving Paradigms of Recombinant Protein Production in Pharmaceutical Industry: A Rigorous Review. Sci. 2024; 6: 9.
- Lakshmanan M, Kok YJ, Lee AP, Kyriakopoulos S, Lim HL, Teo G, Poh SL, Tang WQ, Hong J, Tan AH, Bi X, Ho YS, Zhang P, Ng SK, Lee DY. Multi-omics profiling of CHO parental hosts reveals cell line-specific variations in bioprocessing traits. Biotechnol Bioeng. 2019; 116(9): 2117-2129.
- Zhang JH, Shan LL, Liang F, Du CY, Li JJ. Strategies and Considerations for Improving Recombinant Antibody Production and Quality in Chinese Hamster Ovary Cells. Front Bioeng Biotechnol. 2022; 10: 856049.
- Yang W, Zhang J, Xiao Y, Li W, Wang T. Screening Strategies for High-Yield Chinese Hamster Ovary Cell Clones. Front Bioeng Biotechnol. 2022; 10: 858478.
- Ronda C, Pedersen LE, Hansen HG, Kallehauge TB, Betenbaugh MJ, Nielsen AT, Kildegaard HF. Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol Bioeng. 2014; 111(8): 1604-1616.
- Sun T, Li C, HanL, Jiang H, Xie Y, Zhang B, Qian X, Lu H, Zhu J. Functional knockout of FUT8 in Chinese hamster ovary cells using CRISPR/Cas9 to produce a defucosylated antibody. Eng. Life Sci. 2015; 15: 660-666.
- Rahimi A, Karimipoor M, Mahdian R, Alipour A, Hosseini S, Mohammadi M, Kaghazian H, Abbasi A, Shahsavarani H, Shokrgozar MA. Efficient CRISPR/Cas9-Mediated BAX Gene Ablation in CHO Cells To Impair Apoptosis and Enhance Recombinant Protein Production. Iran J Biotechnol. 2023; 21(2) :e3388.
- Cohen MJ, Chirico WJ, Lipke PN. Through the back door: Unconventional protein secretion. Cell Surf. 2020; 6: 100045.
- Tripathi NK, Shrivastava A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front Bioeng Biotechnol. 2019; 7: 420.
- Saha D, Das PK, Veeranki VD. Developments in the Production of Recombinant Monoclonal Antibodies. Biotechnol J. 2025; 20(10): e70127.
- Cai K, Anderson J, Utiger E, Ferreira G. Viral clearance capability of monoclonal antibody purification. Biologicals. 2024; 85: 101751.
- Schmitz F, Kruse T, Minceva M, Kampmann M. Integrated double flow-through purification of monoclonal antibodies using membrane adsorbers and single-pass tangential flow filtration. Biochem. Eng. J. 2023; 195: 108913.
- Beck A, Nowak C, Meshulam D, Reynolds K, Chen D, Pacardo DB, Nicholls SB, Carven GJ, Gu Z, Fang J, Wang D, Katiyar A, Xiang T, Liu H. Risk-Based Control Strategies of Recombinant Monoclonal Antibody Charge Variants. Antibodies (Basel). 2022; 11(4): 73.
- Kiyoshi M, Oyama T, Shibata H, Suzuki S, Higuchi Y, Tsumoto K, Ishii-Watabe A. The Structural Fingerprint of Therapeutic Monoclonal Antibodies Determined Using a Combination of Near-UV Circular Dichroism and Statistical Approach for Comparative Analysis. Anal Chem. 2025; 97(24): 12578-12586.
- Chen Z, Zeng M, Park SJ, Balakrishnan G, Zhou K, Pan D, Das TK. Bridging size and charge variants of a therapeutic monoclonal antibody by two-dimensional liquid chromatography. J Pharm Biomed Anal. 2020; 183: 113178.
- Senini I, Tengattini S, Rinaldi F, Massolini G, Gstöttner C, Reusch D, Donini M, Marusic C, van Veelen PA, Domínguez-Vega E, Wuhrer M, Temporini C, Nicolardi S. Direct glycosylation analysis of intact monoclonal antibodies combining ESI MS of glycoforms and MALDI-in source decay MS of glycan fragments. Commun Chem. 2024; 7(1): 203.
- Miller ML, Finn OJ. Flow cytometry-based assessment of direct-targeting anti-cancer antibody immune effector functions. Methods Enzymol. 2020; 632: 431-456.
- Nupur N, Chhabra N, Dash R, Rathore AS. Assessment of structural and functional similarity of biosimilar products: Rituximab as a case study. MAbs. 2018; 10(1): 143-158.
- ICH Q10 Pharmaceutical quality system. 2008.
- ICH Q9 Quality risk management. 2023.
- ICH Q5D Derivation and characterisation of cell substrates used for production of biotechnological/biological products. 1998.
- ICH Q13 Guideline on continuous manufacturing of drug substances and drug products. 2023.