Therapeutic Protein Production in Modern Biologic Medicines
- Therapeutic protein production is increasingly viewed not as a fixed set of technologies but as an adaptable, risk-based framework tailored to molecular complexity and intended clinical application.
- Modern therapeutic protein production is dominated by mammalian cell expression systems, especially CHO cell lines.
- Upstream and downstream process development must be co-designed to control impurity profiles and product heterogeneity.
Therapeutic Protein Production in Modern Biologics
The commercial and clinical rise of protein therapeutics has been propelled by platform-like manufacturability for certain classes, especially IgG monoclonal antibodies, and by the expanding range of diseases accessible to large molecules. Therapeutic proteins production includes antibodies, enzymes, cytokines, and hormones. Their pharmacological roles range from neutralization of soluble mediators to replacement of missing enzymes or hormones. The expansion of monoclonal antibodies as a dominant therapeutic class has strongly shaped protein therapeutics manufacturing infrastructure. Antibody-based products now account for a substantial proportion of global biopharma revenues. Their manufacturing paradigms have established industry standards for upstream development and downstream purification platform approaches.
A useful way to connect modality to manufacturing is to recognize that each class couples a characteristic structural risk profile to a characteristic process risk profile. The modular structure of IgG molecules enables relatively standardized purification workflows based on affinity chromatography, while advances in fed-batch and perfusion cell culture have driven volumetric productivity to historically unprecedented levels. At the same time, emerging formats such as bispecific antibodies, antibody-drug conjugates (ADCs), and Fc-fusion proteins introduce new molecular heterogeneity that challenges conventional control strategies.1
From an economic and strategic perspective, biologics manufacturing has become central to the competitive positioning of pharmaceutical companies and contract development and manufacturing organizations. High capital investment requirements for large-scale bioreactor facilities, coupled with stringent GMP protein manufacturing standards, create significant barriers to entry but also incentivize technological innovation.
Expression Systems Used in Therapeutic Protein Production
Expression-system choice is the first major manufacturing commitment in biologics manufacturing. It fixes the feasible envelope for post-translational modifications, impurity profiles, and scale economics. Mammalian cell expression systems dominate production of complex recombinant therapeutic proteins that require human-like glycosylation and correct disulfide pairing, and they do so with a deep regulatory track record that reduces development risk. Reviews of industrial practice describe CHO as the manufacturing host for a substantial majority of marketed protein pharmaceuticals, reflecting a balance of robust growth, adaptability to serum-free suspension culture, and controllable product quality. This dominance is reinforced by the fact that Fc glycosylation affects effector function and pharmacokinetics for antibodies and Fc-fusion proteins, making mammalian processing often the lowest-risk route to clinically acceptable glycoforms.3
Newer modalities sharpen the consequences of expression-system choice in therapeutic protein production. They have to combine structural complexity with tighter clinical expectations for safety and reproducibility. ADCs often begin with a mammalian-expressed antibody intermediate. Chemical bioconjugation steps create additional heterogeneity level and new critical quality attributes related to drug-to-antibody ratio (DAR) and conjugation sites. Fc-fusion proteins use the Fc region to exploit FcRn-mediated recycling and extend serum half-life, which increases the importance of Fc structural integrity, glycosylation, and charge-variant control.4
Upstream and Downstream Processes in Therapeutic Protein Manufacturing
Upstream development translates genetic constructs into a controlled bioreactor environment where cell physiology, productivity, and product quality co-evolve across the culture trajectory. Small-scale pilot studies using microtiter plates and 10L bioreactors are widely used to select proper media, buffers, and environmental setpoints. Large-scale optimization is too costly and slow to serve as the primary experimentation venue.5
High-throughput process development has therefore become a core competency, enabling parallelized exploration of design spaces for bioreactor parameters, and feeding regimens that influence both titer and quality. Bioreactor technology mode selection (batch, fed-batch, or perfusion) frames the dominant process risks. Importantly, upstream optimization is no longer judged only by grams per liter. It is judged by its ability to produce a consistent distribution of glycoforms, charge variants, and aggregate precursors that downstream can reliably remove or control.6

Downstream development is tasked with converting a complex, variable harvest into a drug substance in therapeutic protein production. The final drug have to meet purity, safety, and potency expectations while preserving the molecule’s functional structure. For monoclonal antibodies, Protein A affinity capture remains a cornerstone because it provides high selectivity and strong impurity reduction early in the train. Ion exchange chromatography, hydrophobic chromatography, and size exclusion chromatography are frequently combined with ultrafiltration/diafiltration to remove host-cell proteins, DNA, aggregates, and process-derived impurities, while achieving concentration and buffer exchange targets. Viral inactivation and virus filtration steps are typically integrated for mammalian-derived products to support viral safety regulatory expectations. The robustness of these unit operations must be demonstrated across realistic operating ranges. Because chromatography capacity and buffer consumption can become cost drivers as titers rise, contemporary DSP development also evaluates membrane adsorbers, mixed-mode options, and intensified polishing concepts that preserve clearance while reducing footprint.7
The most consequential shift in modern biologics process development is the recognition that upstream and downstream are a coupled system, and that optimization must occur at the interface rather than within isolated silos. Upstream conditions that suppress problematic host-cell proteins or reduce aggregate formation can simplify DSP and improve robustness, while DSP capabilities and constraints can shape upstream targets for harvest quality and impurity burdens. This coupling becomes even more important as continuous bioprocessing expands to an innovative operational strategy.8
Critical Quality Attributes in Therapeutic Protein Production
Critical Quality Attributes (CQAs) in therapeutic protein production are the measurable properties that must be controlled to ensure the product performs safely and effectively, and they provide the bridge between molecular science and manufacturing practice. Analytical control strategies must be designed to characterize and monitor intended structure and inevitable heterogeneity. Biotech companies for these reasons using orthogonal methods that reduce the chance of blind spots.9
| Critical Quality Attribute | Analytical methods | Description |
| Molecular mass | LC-MS/MS MALDI-TOF-MS Subunit Mass Analysis Native MS | The measured intact mass confirms the expected protein composition. Deviations can indicate truncations, incorrect processing, or unexpected PTMs. |
| Primary amino acid sequence integrity | LC-MS/MS Edman Degradation | The correct primary sequence is foundational to potency and safety because even single-residue changes can alter binding, activity, or immunogenicity risk. Sequence errors may arise from mistranslation, mutations in the expression construct, or proteolytic processing. |
| Higher-order structure | Circular Dichroism Hydrogen–Deuterium exchange MS (HDX-MS) Differential scanning calorimetry (DSC) | Protein folding governs receptor binding, and stability. Secondary structure can be perturbed by process stress or formulation conditions. Subtle conformational changes may affect potency and immunogenicity. |
| Disulfide bond pairing and free thiols | Free thiol quantitation (Ellman’s/ThioGlo) Non-reducing CE-SDS | Correct disulfide connectivity stabilizes higher-order structure and is often required for full biological activity. Mispaired disulfides or elevated free thiols can promote aggregation and reduce stability. |
| Aggregation level | SEC-HPLC SDS-PAGE SV-AUC Dynamic Light Scattering | Aggregation involves the unwanted self-association of protein molecules (monomers, dimers, sub-visible particles). Aggregation propensity is influenced by upstream stress, purification conditions, and formulation/excipient choices. |
| Glycosylation profile | HILIC-UHPLC-FLD LC-MS/MS RP-HPLC-MS MALDI-TOF-MS | N-glycan structures affect stability, Fc effector function, serum half-life, and immunogenicity, making them central CQAs for many biologics. Glycosylation is sensitive to host cell line, media, and culture conditions. |
| Oxidation | Peptide mapping LC-MS/MS Reversed-phase HPLC of oxidized variants | Oxidation can reduce binding affinity, destabilize higher-order structure, and accelerate aggregation, particularly in Fc regions or CDRs. It may be promoted by light exposure, trace metals, or peroxide impurities in excipients. |
| Host cell protein (HCP) residuals | ELISA LC-MS | HCPs can pose immunogenicity risk, or catalyze degradation pathways. Their identities and levels can be process-dependent. |
| Residual host cell DNA | qPCR PicoGreen Fluorescence Assay | Residual DNA is a safety-related impurity. DNA levels are sensitive to harvest clarification efficiency and chromatography performance. |
| Biological activity (potency) | Cell-based potency assay (CDC, ADCC) Surface Plasmon Resonance ELISA | The appropriate potency assay format depends on the molecule and may require both binding and functional readouts. |
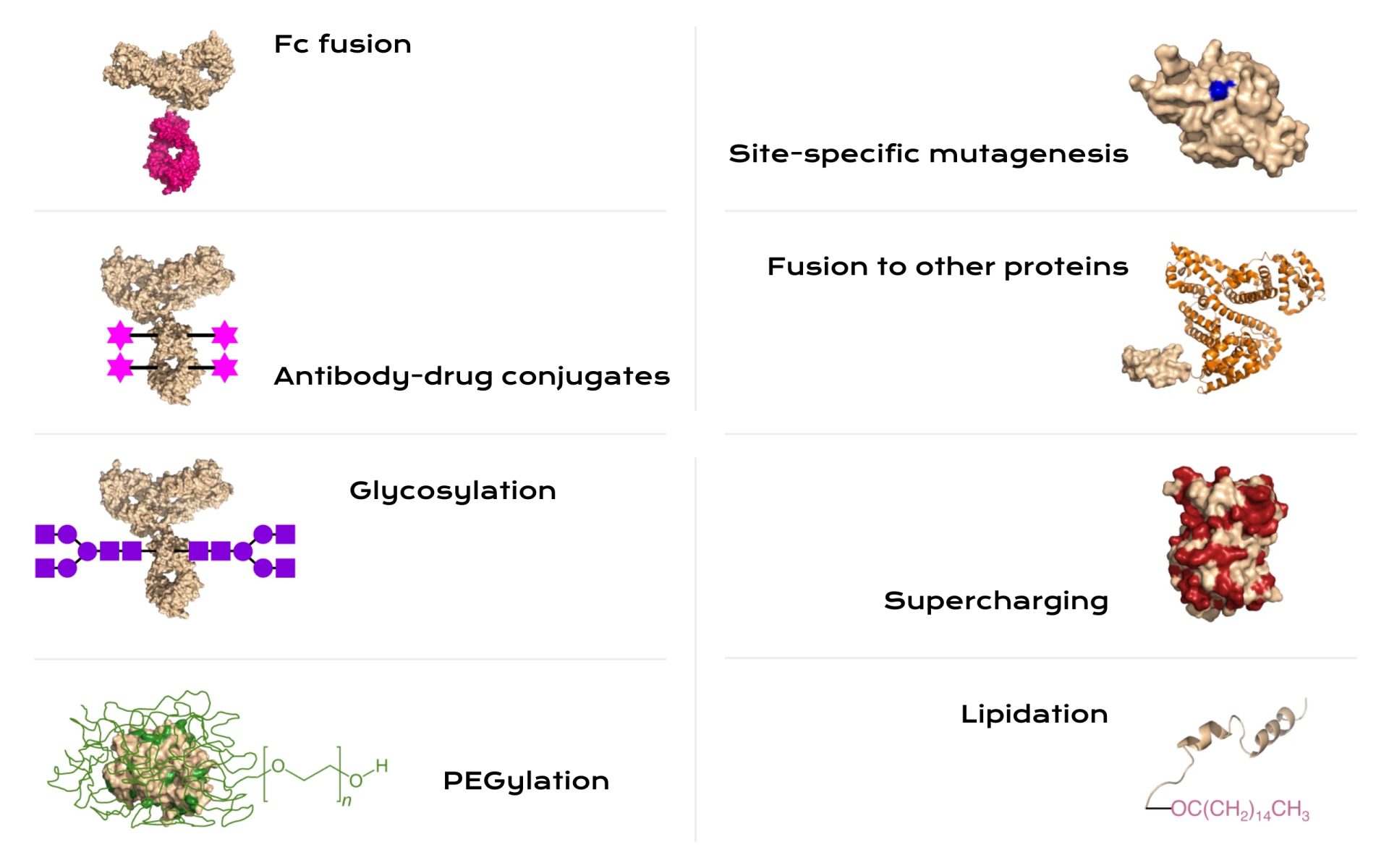
The design of protein-based therapeutics further complicates CQA control. Chemical and structural modifications intended to improve stability, pharmacokinetics, or targeting can introduce new heterogeneity and new failure modes. Strategies such as PEGylation, lipidation, engineered glycosylation, or protein fusion can extend half-life or improve distribution. But they can also change potency, alter immunogenicity risk, or increase sensitivity to certain stresses during storage and handling. Manufacturing must anticipate the downstream consequences of design choices. This is why therapeutic protein production often benefits from an explicit process–structure–function narrative that ties each critical process parameter to a plausible mechanism of action affecting a CQA. In practical terms, the best control strategies treat CQAs as the primary outcomes and view upstream and downstream parameters as levers to shape those outcomes within a validated design space.10,11
GMP and Regulatory Requirements for Therapeutic Protein Manufacturing
GMP-compliant protein manufacturing establishes the regulatory operational foundation for the production of therapeutic proteins. These regulations define validated processes, qualified equipment, controlled environments, and rigorous documentation, which together reduce the likelihood of uncontrolled variability. Regulatory expectations also include data integrity, deviation investigations, and change control.
Internationally harmonized quality frameworks shape how manufacturers translate scientific understanding into regulatory submissions and post-approval lifecycle management.
- ICH Q6B scientific guideline provides expectations for specifications and analytical approaches tailored to biotechnological/biological products.12
- ICH Q5E scientific guideline provides principles for demonstrating comparability when manufacturing changes occur, an inevitability as processes scale-up.13
- ICH Q10 scientific guideline frames a pharmaceutical quality system that links development knowledge to manufacturing execution and encourages continual improvement.14
This posture has practical consequences for biologics manufacturing. It also underscores why process development must be accompanied by an increasingly sophisticated analytical program capable of detecting small but potentially meaningful differences.
Ensuring Consistency and Scalability in Therapeutic Protein Production
Scale-up from bench to pilot to commercial volumes must preserve key transport and mixing phenomena that influence cell physiology, including oxygen transfer, CO2 stripping, shear exposure, and nutrient gradients. Even when average setpoints match, large-scale bioreactors can introduce microenvironments that shift metabolism. Thereby alter biologics glycosylation patterns or increase aggregation precursors, creating downstream burdens that were absent at small scale. This is why robust scale-down models are central to biologics process development and therapeutic protein production. They allow systematic testing of parameter excursions and failure scenarios under controlled conditions before risk is taken at manufacturing scale. The goal is faithful reproduction of the features that matter for CQAs and process capability.
The adoption of platform manufacturing approaches has contributed significantly to improved scalability in biologics manufacturing. Standardized cell culture media, feeding strategies, and purification templates allow organizations to leverage prior knowledge across multiple recombinant therapeutic proteins. Such platform strategies reduce development timelines and facilitate regulatory review by demonstrating consistency in control strategies. However, molecule-specific characteristics must still be carefully evaluated to ensure that platform assumptions remain valid. Continuous improvement initiatives enhance process capability and reduce variability.15
Ensuring consistent therapeutic protein production requires an integrated strategy encompassing cell line stability, process control, analytical rigor, and organizational governance. Cross-functional collaboration is essential to maintain alignment between scientific understanding and operational execution.
FAQ
Prepared by:

Marketing Specialist

Junior Downstream Process Specialist
References
- Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008; 7(1): 21-39.
- Ebrahimi SB, Samanta D. Engineering protein-based therapeutics through structural and chemical design. Nat Commun. 2023; 14(1): 2411.
- Tripathi NK, Shrivastava A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front Bioeng Biotechnol. 2019; 7: 420.
- Yang CW, Xia T, Tan Q, Jie LG, Lou AJ, Li XX, Maitiyaer M, Huang WH, Zheng Y, Yu SL. From promise to practice: evaluating the clinical impact of FcRn inhibition in IgG-mediated autoimmune rheumatic diseases. Front Immunol. 2025; 16: 1656937.
- Monteiro M, Fadda S, Kontoravdi C. Towards advanced bioprocess optimization: A multiscale modelling approach. Comput Struct Biotechnol J. 2023; 21: 3639-3655.
- Rojewska O, Tęczar M. Bioprocess Operation Modes and Advanced Bioreactor Technology. Mabion Science Hub. 2025.
- Ranbhor R. Advancing Monoclonal Antibody Manufacturing: Process Optimization, Cost Reduction Strategies, and Emerging Technologies. Biologics. 2025; 19: 177-187.
- Knurek J. From Research to Release: Mapping Mabion’s Full Analytical Lifecycle for Monoclonal Antibodies. Mabion Science Hub. 2025.
- Metzendorf NG, Petersen I, Hultqvist G. Enhancing therapeutic antibody profiling: orthogonal strategies for stability and quality assessment. Front Pharmacol. 2025; 16: 1667210.
- Kendrick BS, Sampathkumar K, Gabrielson JP, Ren D. Analytical control strategy for biologics. Part I: Foundations. J Pharm Sci. 2025; 114(7): 103826.
- Sampathkumar K, Kendrick BS, Gabrielson JP, Ren D. Analytical control strategy for biologics. Part II: Roadmap for development and implementation. J Pharm Sci. 2025; 114(8): 103834.
- ICH Q6B Specifications: test procedures and acceptance criteria for biotechnological/biological products. 1999.
- ICH Q5E Biotechnological/biological products subject to changes in their manufacturing process: comparability of biotechnological/biological products. 2005.
- ICH Q10 Pharmaceutical quality system. 2008.
- Shukla AA, Wolfe LS, Mostafa SS, Norman C. Evolving trends in mAb production processes. Bioeng Transl Med. 2017; 2(1): 58-69.